
西北农林科技大学小麦多组学数据库的构建与应用
2024-07-07 17:27:49路田刘子辉赵鹏邱四春王晓明郑炜君韩德俊毛虎德宋卫宁陈新宏奚亚军王中华吉万全康振生许盛宝
西北农业学报 2024年6期
路田 刘子辉 赵鹏 邱四春 王晓明 郑炜君 韩德俊 毛虎德 宋卫宁 陈新宏 奚亚军 王中华 吉万全 康振生 许盛宝
摘 要 本研究在大规模根系转录组分析的基础上,获得406份全球遗传多样性丰富的小麦种质的基因型数据,并通过对18个农艺性状在10个种植环境中的系统调查,获得对应的表型数据。实验室前期评估证实表型与基因型数据可靠,可应用于小麦研究。同时,整合基因型、转录组、表型与栽培环境的气象数据,建立了一个可在网站(https://iwheat.net/resource)自由下载与利用的小麦多组学数据库,为小麦功能基因的挖掘与机理研究提供支持。
关键词 小麦;多组学;数据库;基因型;表现型
小麦是全球最重要的粮食作物之一,是人类的主要营养来源[1]。在人口规模快速增长的大背景下,增加小麦产量是当今农业领域最重要的课题之一。开发高产、优质、适应性强的小麦新品种是保障小麦持续供应的必要途径。变异在植物进化过程中始终持续发生,植物通过变异选择适应环境的优良性状稳定遗传,解析变异信息,是研究植物性状遗传的重要手段[2]。随着测序技术的不断发展和进步,研究人员得到了大量的变异数据,为了便于应用这些数据,各种网络共享数据库应运而生。目前,有许多大型公共网站可以免费使用,例如NCBI (National Center for Biotechnology Information)、Uniprot (Universal Protein)等。
随着小麦基因组的快速发展,不同小麦品种间的基因型变异也逐渐被揭示。国际小麦基因组测序联盟(International Wheat Genome Sequencing Consortium,IWGSC)2018年公布了六倍体小麦的最新参考基因组[3],此后小麦的测序数据呈现爆炸式增长,小麦基因组信息可以方便地从国内外的众多公共数据库获得。如,IWGSC数据库包含完整的中国春参考基因组和基因注释信息,Wheat Expression Browser(www.wheat-expression.com)数据库包含大量六倍体小麦基因表达量数据,CerealsDB(www.cerealsdb.uk.net)数据库包含大量六倍体小麦SNP和基因分型数据[4],这两个数据库都很好地提供了大量分子标记,为育种工作提供了便利;WGVD小麦基因组变异数据库包含7 346 814个SNP和 1 044 400个indels[5],该数据库还可以进行可视化基因组变异,并可以利用比对工具Blast来搜索序列之间的同源区域;WheatQTLdb提供了小麦高产、抗逆等多方面研究中已报道的位点与基因座信息[6],其包含小麦的形态学性状、对胁迫的耐受性、质量性状和产量及其相关性状已经报道的位点或基因座;然而这些数据库并没有提供相应的表型数据。此外,还有大量可视化数据库,例如Triticeae-GeneTribe[7]、Snphub[8]与WheatExp[9]等,都可以较为容易地获得小麦基因组、基因表达信息以及不同小麦品种的单倍型信息,这些小麦基因组数据共享为小麦优异基因的发掘提供了坚实的基础。
然而,在基因组学数据库爆炸增长的同时,表型组学数据库的发展较为滞后。目前存在的几个作物表型数据库,如,Planteome[10]和PGP repository[11]都是比较成熟的大型植物表型组学数据发布平台,但其中并没有系统的小麦表型组数据。OPTIMAS-DW数据库提供了玉米的转录组学与表型组学综合数据资源[12];BIOGEN BASE-CASSAVA是一个木薯表型组与基因组信息资源库[13]。多组学数据库的诞生为基础植物研究与作物育种提供了很大帮助,然而目前并没有一个真正成熟的小麦表型组数据库,这成为当前小麦功能基因发掘中的一个短板[14]。因此,构建一个包含小麦基因组与表型组数据的多组学数据库,对于小麦领域研究人员具有重要意义。
本研究利用遗传多样性丰富的406份小麦种质,通过大规模根系转录组分析,获得了406份小麦的基因型数据。在10个不同种植环境下对这些种质的18个农艺性状表型进行系统的观察与记录,得到了一份与基因组对应的表型组数据,同时记录了10个栽培环境的气象数据。经过实验室前期的应用与评估,表型与基因型数据良好可靠,可以应用到小麦研究的多个方面。该数据库包含了表型、基因型以及对应的气象数据,希望能给小麦领域研究人员提供一些研究便利。
1 材料与方法
1.1 试验材料的收集
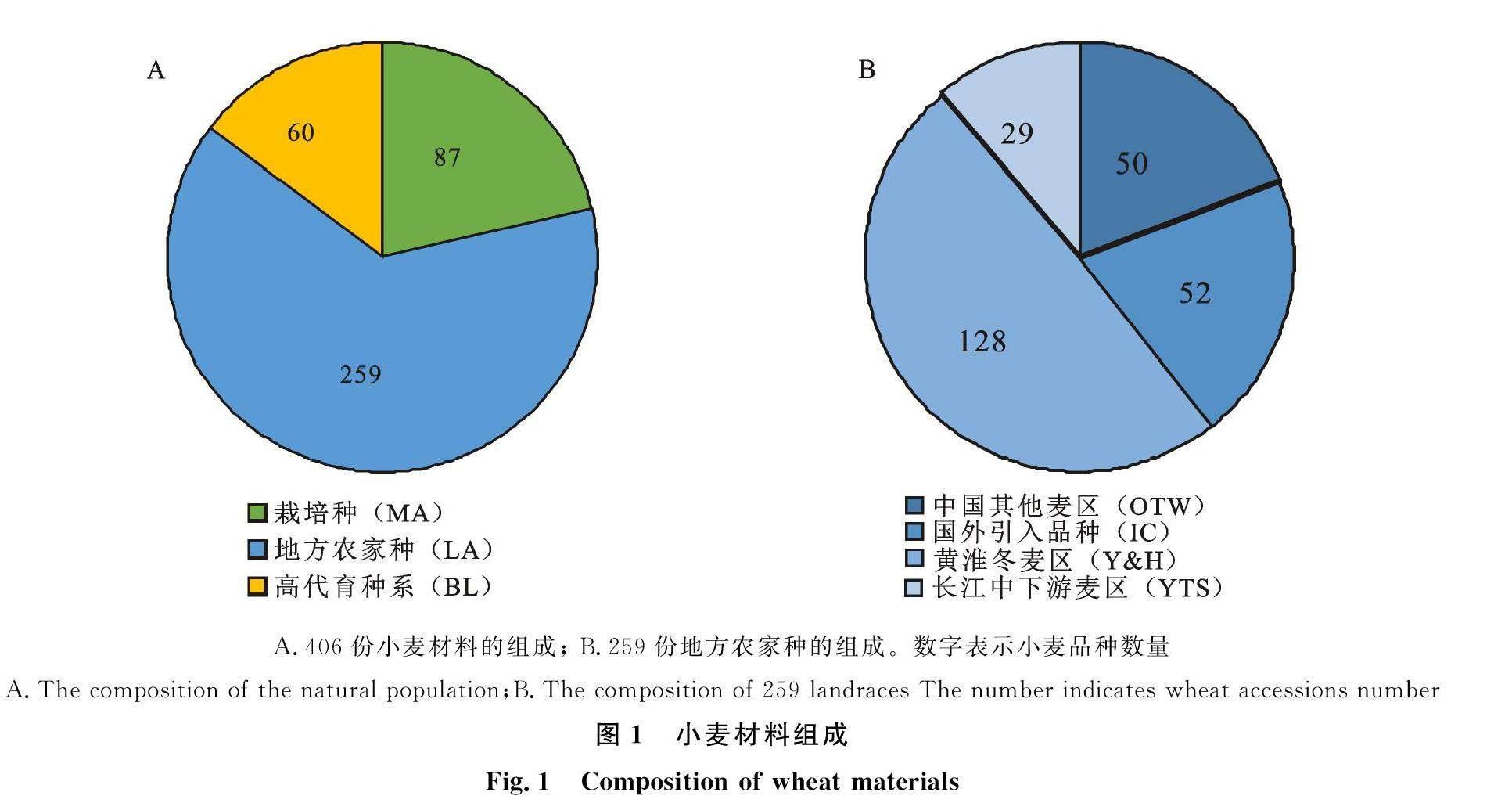
供试材料为亲缘关系彼此较远的406份小麦种质资源,其中包括87份地方农家种,259份栽培种,60份高代育种系。在259份栽培种中,包括205份中国小麦现代品种(128份来自中国黄淮冬麦区,29份来自长江中下游冬麦区,18份来自北方冬麦区,15份来自西南春麦区,8份来自东北春麦区,2份来自青藏春冬兼播麦区,2份来自西北春麦区,1份来自新疆冬春兼播麦区,2份材料不能确定具体麦区)与52份国外引进的品种(9份来自意大利;6份来自加拿大;4份来自美国;3份来自智利;以及来自德国、巴西、南非的各2份材料;来自阿根廷、埃及、澳大利亚、法国、哥伦比亚、摩洛哥、葡萄牙、日本、塞尔维亚、伊朗、以色列、印度、英国的各1份材料,11份材料不能确定具体国家);60份高代育种系分别来自从国际旱地农业研究中心ICARDA引进的小麦抗逆材料,以及从美国引进的高代育种系材料。
1.2 田间表型测定
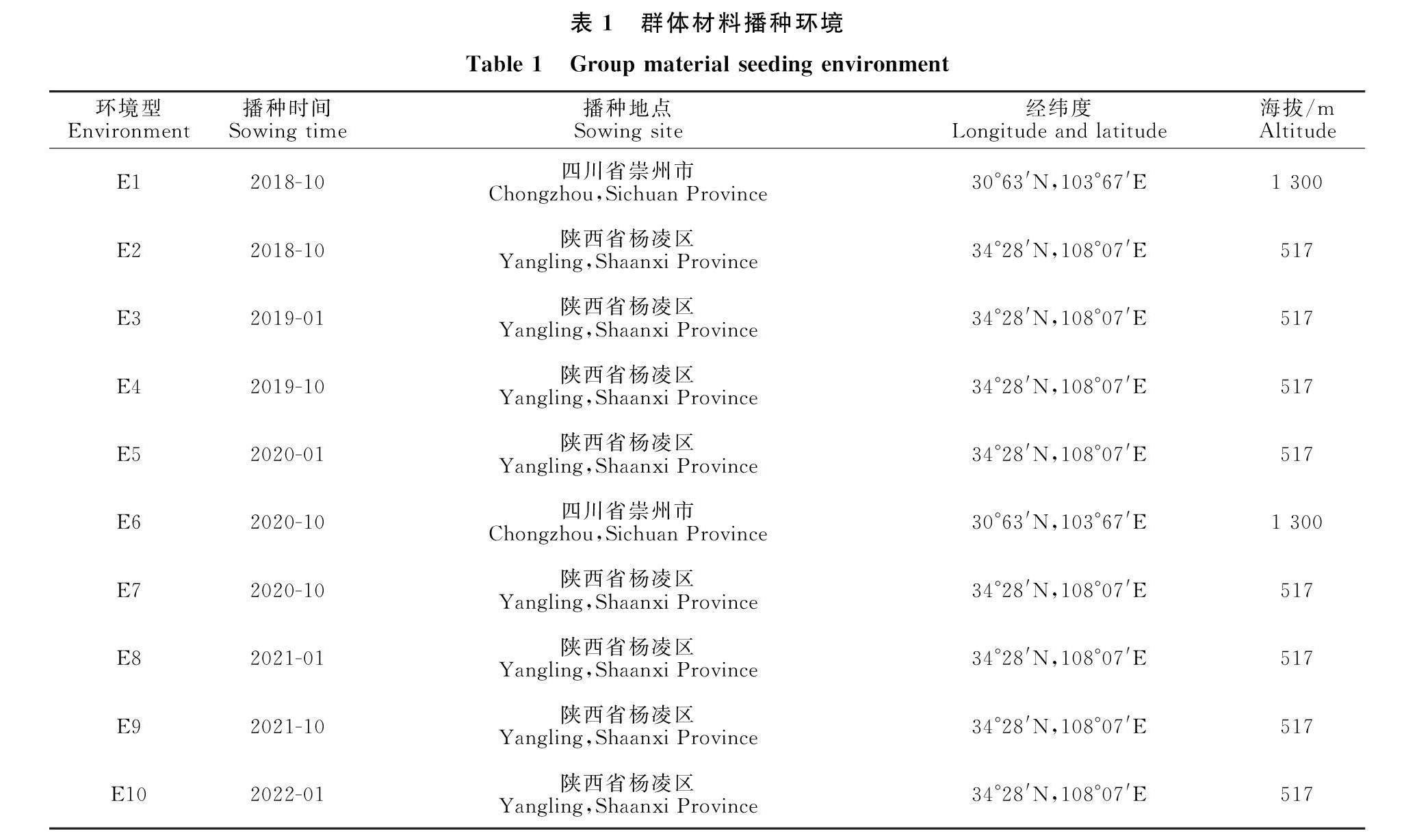
1.2.1 田间试验 在陕西省咸阳市杨陵区曹新庄试验农场于2018-2019、2019-2020和 2020-2021、2021-2022年当年10月初正常播种和次年1月中旬进行晚播;在四川省崇州市四川农业大学现代农业研究基地于2018-2019年和2020-2021年当年10月末进行正常播种。每个材料3个重复,行长1 m、行距20 cm,每行点播10粒种子,并根据当地生产情况进行田间管理。
1.2.2 农艺性状表型测定 于小麦抽穗期开花期统计每个重复各种质的抽穗期(Heading date)、开花期(Flowering date)、灌浆持续时间(Grain filling date)。
于小麦成熟期人工统计每个重复各种质的株高(Plant height,PH),穗下节长度(Peduncle length,PL)、总分蘖数(Total tiller number,TTN)、有效分蘖数(Productive tiller number,PTN)、旗叶长(Flag leaf length,FLL)、旗叶宽(Flag leaf width,FLW)、旗叶夹角(Flag leaf angle,FLAN)、旗叶面积(Flag leaf area,FLA)[15],每个重复调查3个样品。
每个重复收获5个长势一致的小麦穗子,人工统计穗长(Spike length,SL)、总小穗数(Total spikelet number,TSN)、可育小穗数(Fertile spikelet number,FSN)、穗粒数(Kernel number per spike,KNS)。
利用SC-G谷粒外观质量快速图像分析系统分析每个重复各种质的千粒质量(Thousand kernel mass,TKW)、籽粒长(Kernel length,KL)、籽粒宽(Kernel width,KW)。
1.3 根系表型测定
选取饱满一致的小麦种子,将种子消毒、吸胀,将打破休眠后的种子放入发芽盒中,将406份材料分别于培养箱中培养4 d、7 d、14 d,用直尺测量总根长(Total root length TRL)、主根长(Primary root length PRL);于贴近种子处剪下小麦初生根,擦拭水分,测量根鲜质量(Root fresh mass,RFW)。利用万深LA-S植物根系扫描仪进行根系表型扫描并分析,得到根表面积(Root surface area,RS)和根体积(Root volume,RV)。
1.4 气象数据收集
通过国家气象信息中心(https://data.cma.cn),统计10个环境的最高气温、最低气温、降水量、日照时数、相对湿度和太阳辐射共6个气象因子。
1.5 数据处理
利用Microsoft office EXCEL软件和Python程序语言对得到的10个环境农艺性状、根系表型、气象数据进行处理,利用R语言的R包Lme4计算最佳线性无偏估计值(Best linear unbiased estimate,BLUE)。
1.6 转录组测序
与武汉菲沙公司合作对这406份小麦材料的苗期根系进行了大规模转录组测序(RNA-seq)与组装,对测序得到的数据进行过滤以及质量控制,过滤标准为:只保留纯合基因型,并且每个样本的支持reads必须大于等于30。通过小麦660 K SNP微阵列对过滤到的SNP准确性进行评估[16]。
1.7 在线数据库搭建
本数据库利用R语言Shiny框架进行构建,使用R语言的R包shinydashboard编写UI模块设计web网页的页面前端布局,编写Server模块实现后端程序的功能并且控制输出,将两模块的代码整合,利用R包DT和CSS语言代码块将整合结果输出为网页数据库的外观界面并进行相关属性配置。
2 结果与分析
2.1 数据库群体组成、基因型与表型
本数据库包含406份遗传多样性丰富的小麦种质资源,其中包含87份地方农家种(Landraces,LA),259份栽培种(Modern cultivars,MC),60份高代育种系(Breeding lines,BL)。在259份栽培种中包括128份材料来自黄淮冬麦区(Yellow and Huai wheat zone,Y&H),29份材料来自长江中下游冬麦区(Yangtze River wheat zone,YTS),50份材料来自中国其他麦区(Other wheat zone,OTW),52份材料属于国外引入品种(Introduced abroad modern cultivars,IC)(图1)。
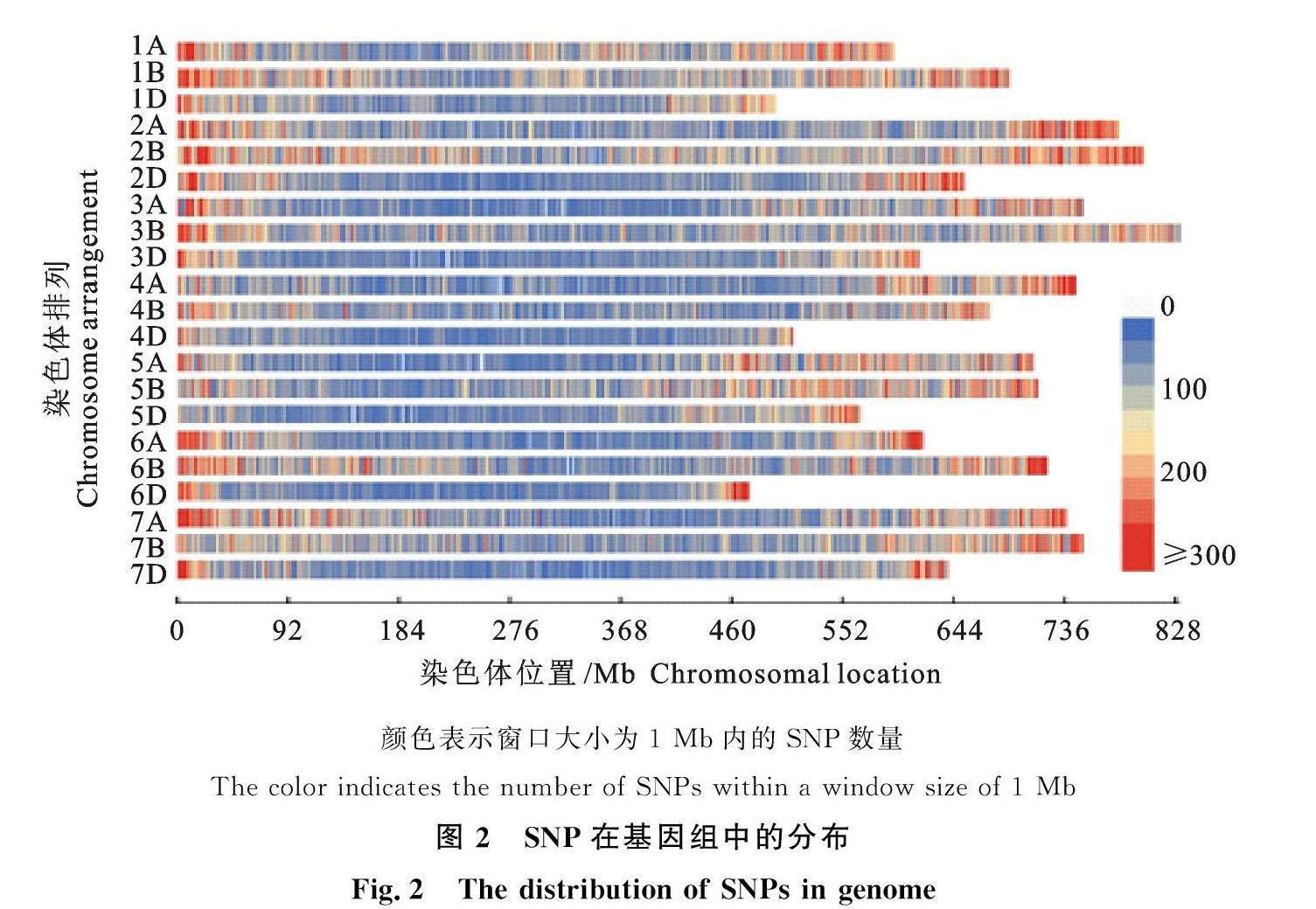
通过与武汉菲沙公司合作对这406份小麦材料的苗期根系进行了大规模转录组测序(RNA-seq)与组装,共鉴定到了1 232 311个单核苷酸多态性(SNP)(图2),利用小麦660 K SNP微阵列评估鉴定SNP准确性>99%,大约一半的SNP(48.43%)位于CDS(编码DNA序列)区域[16]。
同时,通过对这406份材料进行了4 a 10个环境的种植(表1),并且系统调查了18个农艺性状的表型(表2)。同时对4 a 10个环境对应的气象数据进行了收集。数据库也收录了其中351份材料出苗后4 d、7 d,406份材料出苗后14 d的苗期根系表型,此外,利用红外分析仪对每个环境下收获籽粒的品质性状也进行了测量,但使用时发现除蛋白含量这一指标比较稳定外,其余性状在不同环境中的变异非常大,为避免误导他人,这份数据并未提供下载,如有需要,可以单独索取,作为研究参考。
2.2 数据库的使用
本数据库包含6个部分,分别为小麦群体材料信息部分(Sample Information)、基因型数据(Genotypic Data)、基因表达数据(Gene Expression Data)、表型数据(Phenotypic Data)、气象数据(Meteorologic Data)以及小麦群体高质量SNP信息(SNP root and Yield Traits)。
小麦群体材料信息包含406份小麦材料的编号、名称、类型以及来源(图3)。
基因型数据包含406份小麦材料的原始和估算SNP,测序数据映射到IWGSC RefSeq v1.0。可根据不同染色体位置进行下载。基因表达数据包含高置信度和低置信度基因的表达(百万分之一转录本,TPM)。该数据在小麦根系中鉴定出39 243个LC基因,扩展了小麦基因表达数据库。
表型数据包括18个农艺性状与根系表型。农艺性状按照株型性状、产量性状、生育期性状分环境提供,根系性状分别提供了4 d、7 d、14 d的根系表型。可根据需要自行下载(图4)。
气象数据包括7种环境中的最高和最低温度、降水量、日照时数、相对湿度和太阳辐射。可根据需要下载不同环境数据。
高质量SNP标记信息包含SNP所在染色体及物理位置(Chr、Pos)、碱基突变类型(REF、ALT)、对农艺性状和根部表型的影响情况(Down表示下调表型、Up表示上调表型)以及在每个材料的变异情况(0/0表示与参考等位基因型一致,1/1表示与变异等位基因型一致);并提供不同染色体的SNP标记数据(图5)。研究人员可以根据需要进行下载,得到SNP标记信息以及这些标记在群体材料中的分布情况信息。
综上,研究人员通过本数据库获得的信息,可以为后续的育种工作提供参考。
2.3 数据库应用
2.3.1 GWAS的应用
小麦农艺性状的基因定位是小麦研究中的一项重要工作,是发掘小麦优良单倍型与研究基因功能的重要基础。用于QTL定位的群体组成形式多样,主要包括重组自交系、双单倍体系与小麦多品种的自然群体。本数据库包括406份遗传多样性丰富的小麦种质资源以及4 a 10个环境的性状数据,前期用株高数据进行了GWAS评估,发现 Rht-D1b与 Rht-B1b对株高贡献较大,符合长期以来的共识,因此,初步认为该数据库中的表型数据与标记较为可靠。除这两个强效位点之外,利用上述数据同时定位到了多个与株高关联的位点,通过对其中一个位点的验证发现,该位点可以通过控制穗下节来控制株高,同时调整穗下节在全株的比例[17]。
本数据库群体较大,包括406份小麦种质,即使分成两个自然群体,也可以有效进行QTL定位。利用resampling GWAS进行QTL定位,可以组成更多有效的GWAS群体,筛选出潜在位点。据此对所有的18个农艺性状都做了GWAS分析,从运行的结果来看,效果比较理想,可以关联到大量显著标记[18]。本数据库提供了全部个体基因型,可以有根据地组配群体结构,避免群体出现大量标记连锁的不平衡分离现象,为更好选择与组配定位群体提供了更多的选择。
本数据库表型可以作为其他定位结果的重要参考、帮助选择可靠位点并缩小定位区间。本数据库的标记来自大规模的转录组分析,一方面可为精细定位提供更多标记,同时可为确定候选基因提供参考。此外,利用这套数据可评估候选位点对全部18个农艺性状是否具有遗传效应,以评估目标位点的多效性。
2.3.2 不同单倍型在育种中选择与应用的分析 通过查阅记录,逐步理清406份种质资源的品种属性。参考英国John Innes Centre的经验,将1960年前育成或已经种植的小麦材料划分为农家种,而1960年后释放的品种认定为现代育成品种。对于现代品种的栽培地域,限于记录不全与年代久远,部分品种不能确认年代与种植区域,在259份栽培种中,有26份材料没有年代信息,2份材料不能确认地域信息。
根据这些信息可以分析群体中各SNP在现代育种过程中的选择力度,以及不同种植区域对SNP的选择情况。如发现的降低穗下节的单倍型,除降低穗下节外,还能增加穗粒数与粒质量,是个非常有价值的优异基因单倍型。育种选择分析表明这个位点的单倍型在栽培种中被显著选择(图6-A),但这个选择只在中国的现代小麦品种中发生了,而国外品种中却几乎没有被选择利用(图6-B),所以有时会称它为中国单倍型[17]。
2.3.3 探究环境对小麦生长发育及育种改良的影响 小麦作为固着生长的植物,其生长发育受到气象条件的严格调控,小麦的各种农艺性状是基因型与环境因子互作的结果,因此,研究气象条件对小麦生长习性的调控,是高产优质小麦品种选育与栽培的重要基础。然而目前对于小麦响应环境的机理并不清楚。
采用多元回归分析方法研究气象因子对小麦生长发育的影响是小麦育种工作中的常用方法。本数据库收集了小麦群体在不同生长环境中的6个气象因子。结合不同品种在不同环境下发育进程的记录,可以得到不同品种在不同发育时期经历的气象因素变化,通过拟合多元线性回归方程可探究不同环境因子对小麦表型、生育时期的影响。如笔者利用多元回归分析拟合其中7个生长环境的不同气象因子与13个农艺性状的表型数据的方程,发现温度对几乎所有的小麦农艺性状都有显著影响,尤其是株高、分蘖数和千粒质量等[18]。此部分数据有望能为探究环境影响小麦生长发育的科研工作者提供一定数据支持。
2.3.4 不同SNP可能参与的发育过程进行评估
本数据库转录组测序得到的1 232 311个高质量SNP。可以根据任一SNP的不同单倍型将406份材料划分为两个群体,结合多环境的表型,分析该SNP是否显著地改变了小麦农艺性状,初步推断该SNP参与了小麦哪些发育过程,从而为深入研究该SNP所在基因的功能提供切入点与基础。据此本数据库鉴定到了大量对千粒质量、穗粒数、有效分蘖数、根表面积等性状存在潜在影响的高质量SNP。同时,这部分的结果也可以为实验室中研究小麦基因功能提供一个平行的田间表型证据。
3 讨 论
这套表型数据中一共包含了18个田间农艺性状,彼此之间呈现一定的关联性。这些表型相互组合以后又会产生新的内涵,比如,穗长除以每穗小穗数,可以得到穗间距;旗叶长与旗叶宽的比例,可以得到旗叶长宽比,可以拓展到旗叶形状与细胞形状的研究中[19];而把有效分蘖数、穗粒数、粒质量进行组合后,可以进行小麦单株产量的基因定位;而利用单株产量/株高×分蘖数则有助于定位到一些与经济系数相关的位点;同时如果根系里每个表达的基因量也可以作为一个性状的话,控制每个基因表达的位点也有希望能采用GWAS分析去探索。因此,使用者可以根据自己的研究兴趣,利用该数据库做出相应的拓展。
GWAS是一种常用的发掘作物优异基因的方法。近年来已经大量开展了关于GWAS表型分析的流程、模型、方法的探索性研究。如将主成分作为因变量进行全基因组关联分析[20];将不同环境间的方差作为表型进行关联,从而筛选不同环境中稳定出现的候选基因[21-22];使用各环境的表型均值也可以定位到共同稳定的位点,去发现独立于环境的遗传位点[23-24];此外,也可以利用核苷酸多态性的估计效应筛选候选基因[25];目前在小麦中GWAS已逐渐成为一种常用的优异基因发掘的方法,相对于其他物种,小麦的基因组大,冗余性高,对于GWAS的定位易造成假阳性高与定位区间大等限制。多年多点可靠的表型与基因型数据将可以为不同方向的研究人员提供基础数据,希望这份数据能为发展与优化小麦GWAS流程做出一些贡献。此外,根据这套数据也尝试做了TWAS分析,并且找到了Rht-D1b与小麦根系。然而,基于目前小麦的TWAS流程现状,这个结果存在一定的偶然性,尚需该领域的专家学者利用这套数据做更好的评估与开发。
2022年Nature发表了16份小麦的全基因组的序列,为小麦泛基因组正式拉开了序幕[26],结合此前遗传所鲁非的414份小麦的外显子测序[27],农科院张学勇的145份小麦测序[28],以及西农姜雨的93份小麦基因组测序[29],进一步推进了小麦的泛基因组研究。泛基因组指的是同一物种的全部基因包括核心基因组与可变基因组,泛基因组可通过将测序个体与参考基因组比对,获得完整的群体变异情况,很好地解决了传统基因组学研究中,参考基因组不能完全覆盖小麦的全部遗传信息的问题,同时泛基因组是研究不同小麦品种的外源基因渗入、染色体片段的插入易位与重组等信息,以及挖掘小麦稀有单倍型的重要基础,也将是未来小麦基因发掘的重要研究方向。
本项目中的转录组包含406份小麦根系转录组信息,得到了不同品种间的SNP,首先鉴定到的39 243个根系表达的基因是在国际小麦基因组测序联盟(IWGSC)的低可信基因库中(iwgsc_refseqv1.1_genes_2017July06),为这部分基因提供了表达的证据,为研究人员选择基因提供了可选的参考。这些获得的全部集合可以作为小麦苗期根系的泛转录组,尽管不能涵盖小麦的基因组,但其中的外源基因深入、易位与插入等信息可为当前的泛基因组提供重要的补充。可以作为根系泛转录组与小麦泛基因组新的切入点。
经过5 a多的基因型与表型的调查与评估,目前这个包含406份小麦材料的群体展现出可靠与稳健地结果输出能力,可以用于小麦研究的多个方面。旱区逆境作物生物学重点实验室与课题组希望通过分享该数据库,为国内小麦同行提供更多的研究选择,共同推进国内小麦基础与育种研究。同时,在利用的同时希望进一步提升与拓展该群体的质量与应用范围,以期成为广大小麦工作者的一个可靠支撑,因此也欢迎广大同行在使用时,不断提出存在问题与改进建议,共同做好这份数据的持续性升级与利用。
致谢:感谢所有作者在种质收集、种质筛选与种质划分,以及表型调查中的支持与建议。感谢四川农业大学陈国跃与蒲至恩老师表型调查中的支持。
参考文献 Reference:
[1] APPELS R,EVERSOLE K,STEIN N,et al.Shifting the limits in wheat research and breeding using a fully annotated reference genome [J].Science,2018,361:7191.
[2]OTTO S P,GERSTEIN A C.The evolution of haploidy and diploidy [J].Current Biology,2008,18(24):1121-1124.
[3]IWGSC.Shifting the limits in wheat research and breedingusing a fully annotated reference genome [J].Science,2018,361(6403):7191.
[4]WILKINSON P A,ALLEN A M,TYRRELL S,et al.CerealsDB-new tools for the analysis of the wheat genome:update 2020 [J/OL].Database,2020.01.01.
[5]WANG J R,FU W W,WANG R,et al.WGVD:an integrated web-database for wheat genome variation and selective signatures[J/OL].Database,2020.01.01.
[6]SINGH K,BATRA R,SHARMA S,et al.WheatQTLdb:a QTL database for wheat [J].Molecular Genetics And Genomic,2021,296(5):1051-6.
[7]CHEN Y,SONG W,XIE X,et al.A collinearity-incorporating homology inference strategy for connecting emerging assemblies in the triticeae tribe as a pilot practice in the plant pangenomic era [J].Molecular Plant,2020, 13(12):1694-708.
[8]WANG W,WANG Z,LI X,et al.SnpHub:an easy-to-set-up web server framework for exploring large-scale genomic variation data in the post-genomic era with applications in wheat [J].Gigascience,2020,9(6):1-8.
[9]PEARCE S,VAZQUEZ-GROSS H,HERIN S Y,et al.WheatExp:an RNA-seq expression database for polyploid wheat [J].BMC Plant Biology,2015,15:299.
[10] COOPER L,MEIER A,LAPORTE M A,et al.The Planteome database:an integrated resource for reference ontologies,plant genomics and phenomics [J].Nucleic Acids Research,2018,46(D1):1168-1180.
[11]AREND D,JUNKER A,SCHOLZ U,et al.PGP repository:a plant phenomics and genomics data publication infrastructure[J/OL].Database,2016.04.17.
[12]COLMSEE C,MASCHER M,CZAUDERNA T,et al.OPTIMAS-DW:a comprehensive transcriptomics,metabolomics,ionomics,proteomics and phenomics data resource for maize[J].BMC Plant Biology,2012,12:245.
[13]JAYAKODI M,SELVAN G,NATESAN S,et al.A web accessible resource for investigating cassava phenomics and genomics information:BIOGEN BASE [J].Bioinformation,2011,6(10):391-392.
[14]王璟璐,张 颖,潘晓迪,等.作物表型组数据库研究进展及展望 [J].中国农业信息,2018,30(5):13-23.
WANG J L,ZHANG Y,PAN X D,et al.Research progress and prospect on cropphenomics database[J].China Agricultural Informatics,2018,30(5):13-23
[15]FAN X,CUI F,ZHAO C,et al.QTLs for flag leaf size and their influence on yield-related traits in wheat (Triticum aestivum L.)[J].Molecular Breeding,2015,35:1-16.
[16]WANG X,ZHAO P,GUO X,et al.Population Transcriptome and Phenotype Reveal that the Rht-D1b Contribute a Larger Seedling Roots to Modern Wheat Cultivars [J].BioRxiv,2022.Doi:https://doi.org/10.1101/2022.06.02.494553.
[17]LIU Z,ZHAO P,LAI X,et al.The selection and application of peduncle length QTL QPL_6D.1 in modern wheat (Triticum aestivum L.) breeding [J].Theoretical and Applied Genetics,2022,136(3):32.
[18]LIU Z,HU Z,LAI X,et al.Multi-environmental population phenotyping suggests the higher risk of wheat Rht-B1b and Rht-D1b cultivars in global warming scenarios [J/OL].BioRxiv,2022.07.18.
[19]CHEN S,LIU F,WU W,et al.A SNP-based GWAS and functional haplotype-based GWAS of flag leaf-related traits and their influence on the yield of bread wheat (Triticum aestivum L.) [J].Theoretical and Applied Genetics,2021,134(12):3895-3909.
[20]YANO K,MORINAKA Y,WANG F,et al.GWAS with principal component analysis identifies a gene comprehensively controlling rice architecture [J].Proceedings of The National Academy of Sciences of The United States of America,2019,116(42):21262-21267.
[21]RIEDELSHEIMER C,LISEC J,CZEDIK-EYSENBERG A,et al.Genome-wide association mapping of leaf metabolic profiles for dissecting complex traits in maize [J].Proceedings of The National Academy of Sciences of The United States of America,2012,109(23):8872-8877.
[22]苏丽娜.玉米苞叶表型可塑性的GWAS分析 [D].沈阳:沈阳农业大学,2019.
SU L N.GWAS analysis of phenotypic plasticity of maize bracts [D].Shenyang:Shenyang Agricultural University,2019.
[23]WU J,YU R,WANG H,et al.A large-scale genomic association analysis identifies the candidate causal genes conferring stripe rust resistance under multiple field environments [J].Plant Biotechnology Journal,2021,19(1):177-191.
[24]LOU H,ZHANG R,LIU Y,et al.Genome-wide association study of six quality-related traits in common wheat (Triticum aestivum L.) under two sowing conditions [J].Theoretical and Applied Genetics,2021,134(1):399-418.
[25]YANO K,YAMAMOTO E,AYA K,et al.Genome-wide association study using whole-genome sequencing rapidly identifies new genes influencing agronomic traits in rice [J].Nature Genetics,2016,48(8):927-934.
[26]WALKOWIAK S,GAO L,MONAT C,et al.Multiple wheat genomes reveal global variation in modern breeding [J].Nature,2020,588(7837):277-283.
[27]ZHOU Y,ZHAO X,LI Y,et al.Triticum population sequencing provides insights into wheat adaptation [J].Nature Genetics,2020,52(12):1412-1422.
[28]HAO C,JIAO C,HOU J,et al.Resequencing of 145 landmark cultivars reveals asymmetric sub-genome selection and strong founder genotype effects on wheat breeding in China [J].Molecular Plant,2020,13(12):1733-1751.
[29]CHENG H,LIU J,WEN J,et al.Frequent intra- and inter-species introgression shapes the landscape of genetic variation in bread wheat [J].Genome Biology,2019, 20(1):136.
Construction and Application of Wheat Multiomics Database at Northwest A&F University
LU Tian1,2,LIU Zihui1,2,ZHAO Peng1,2,QIU Sichun1,2,WANG Xiaoming1,2,
ZHENG Weijun1,2,HAN Dejun1,2,MAO Hude1,2,SONG Weining1,2,
CHEN Xinhong1,2,XI Yajun1,2,WANG Zhonghua1,2,JI Wanquan1,2,KANG Zhensheng2,3 and XU Shengbao1,2
(1.College of Agronomy(Academy of Agricultural Sciences),Northwest A&F university,Yangling Shaanxi 712100,China;
2.State Key Laboratory of Crop Stress Biology for Arid Areas,Northwest A&F University,Yangling Shaanxi 712100,China;
3.College of Plant Protection,Northwest A&F University,Yangling Shaanxi 712100,China)
Abstract Based on the analysis of large scale root system transcriptome. 406 wheat genotype data with global genetic diversity were collected. The investigation encompassed 18 agronomic traits across in 10 planting environments. A reliable wheat multi-omics database was created by integrating genotype,transcriptome,phenotype,and meteorological data of planting environments,accessible at https://iwheat.net/resource. The database facilitates functional gene exploration and mechanism research in wheat.
Key words Wheat; Multi-omics; Database; Genotype; Phenotype
Received 2023-01-08 Returned 2023-04-26
Foundation item Regulation of TaGSK on Lateral Root Development in Wheat (No.CSBAA202210).
First author LU Tian,female,master student.Research area:molecular design breeding for wheat stress resistance.E-mail:Lut17@nwafu.edu.cn
Corresponding author XU Shengbao,male,professor,doctoral supervisor.Research area:molecular mechanism of heat resistance in wheat. E-mail: xushb@nwsuaf.edu.cn
(责任编辑:成 敏 Responsible editor:CHENG Min)
猜你喜欢
今日农业(2022年16期)2022-11-09 23:18:44
金桥(2021年10期)2021-11-05 07:23:28
今日农业(2021年13期)2021-08-14 01:38:00
作文小学中年级(2020年4期)2020-06-11 12:47:08
新课程·下旬(2018年9期)2018-11-14 12:49:12
中学生理科应试(2016年7期)2016-05-14 15:39:39
中学生理科应试(2015年12期)2016-01-11 10:34:20
高中生学习·高三版(2015年2期)2016-01-04 22:41:24
现代检验医学杂志(2015年6期)2015-02-06 01:44:02
实验动物与比较医学(2014年5期)2014-02-28 14:53:10
