
基于InceptionV3_SVM 模型的蛋白质-ATP 绑定位点预测
2024-06-26 07:57:45宋泽瑞宋初一宋佳智姜静清
电脑知识与技术 2024年14期
宋泽瑞 宋初一 宋佳智 姜静清


摘要:蛋白质-ATP绑定位点预测作为近年来生物医学领域的热点研究之一,对于医药学的发展极其重要。为了提高蛋白质-ATP绑定位点预测的准确率,提出了一种基于深度卷积神经网络和支持向量机融合(InceptionV3_SVM) 的预测方法。首先对蛋白质序列进行特征提取,再采用InceptionV3模型扩大输入数据感受野,对卷积神经网络提取到的深度特征应用SVM分类器进行训练,得到最终预测结果。实验结果表明,该预测方法能够更精确的识别蛋白质-ATP绑定位点。
关键词: 蛋白质-ATP绑定位点;深度卷积神经网络;蛋白质序列;特征提取;SVM
中图分类号:TP18;Q51 文献标识码:A
文章编号:1009-3044(2024)14-0004-06 开放科学(资源服务)标识码(OSID) :
0 引言
在蛋白质-ATP绑定位点的相关预测研究中,主要预测方法有基于生化实验的生物学方法和基于人工智能的计算预测方法等。其中应用生物学方法已经可以比较准确地识别蛋白质-ATP绑定位点,但其所需要的时间和经济成本相对较大,很难满足当前大规模应用的需求[4]。在计算预测方法中,应用传统机器学习算法预测蛋白质-ATP绑定位点的方法主要有支持向量机(SVM) 、随机森林(RondomForest) 等。2011年CHEN K等人提出了基于位置特异性得分矩阵(PSSM) 序列信息的支持向量机方法[5]、2015年石大宏提出结合加权下采样与基于聚类的下采样分别和支持向量机相结合的方法[6]、2015年余健浩等人采用基于支持向量回归集成的方法[7]、2020年SONG J Z等人使用基于SMOTE算法和随机森林分类器融合的方法[8],分别在蛋白质-ATP绑定位点预测研究中的不平衡数据处理和精度预测等方面做出了相关研究。相比于传统机器学习的预测方法,应用深度学习算法预测蛋白质-ATP绑定位点的方法主要有卷积神经网络(CNN) 、循环神经网络(RNN) 、自编码器等。2019年郭丽萍等人基于自编码器和卷积神经网络构建了深度学习预测模型[9]、2019年张寓等人实现了基于一维卷积神经网络的模型[10]、2022年刘桂霞等人提出了改进的Inception卷积架构的深度网络模型[11],分别利用深度学习算法构建多种网络架构的方式对蛋白质-ATP 绑定位点进行了预测,并且预测结果得到了提升。
以往研究采用的分类算法主要为传统的机器学习算法或深度学习算法,对蛋白质-ATP绑定位点的预测做出了开创性的贡献,但其预测精度仍有进一步提升的可能,对于蛋白质-ATP绑定位点预测研究中的生物数据分析和样本分类预测性能仍有待加强。本文基于改进的深度学习算法InceptionV3卷积网络模型和机器学习算法相结合的思想,提出了Incep?tionV3_SVM分类架构,用以实现蛋白质-ATP绑定位点的预测。最后的实验结果表明,InceptionV3_SVM 分类架构可以有效提升预测方法的整体性能,对于蛋白质和其他配体绑定位点的预测研究同样具有重要意义。
1 数据集及评价指标
1.1 数据集
本文所使用的数据集是ATP-388和ATP-41数据集[12],该数据集共有429条含有ATP绑定位点的蛋白质序列。该数据集来自2016 年之前的PDB(ProteinData Bank) 中的数据记录,通过使用CD-hit软件去除蛋白质记录中同源性超过40% 的冗余序列,最终得到429条非冗余的蛋白质序列数据。在这429条蛋白质序列中,388条蛋白质序列作为训练集,41条蛋白质序列作为独立测试集。
1.2 滑动窗口
研究表明,蛋白质序列中残基的ATP结合特性不仅与其自身理化属性相关,同时也会受到其相邻残基理化属性的影响。因此,一般使用滑动窗口的方法把目标残基和邻近残基的特征值进行整合作为一个目标残基的总特征值。滑动窗口处理过程如图1所示。当滑动窗口大小为L时,一个蛋白质目标残基会整合其前(L-1)/2个相邻残基和后(L-1)/2个相邻残基的特征值作为总特征值,在目标残基前后的相邻残基不足(L-1)/2个时,所缺少的残基特征值全部用0补齐,其处理过程如图1所示。在本文研究中,经过多次实验尝试,当滑动窗口大小L=17时,预测方法可以获得最优性能。
1.3 特征提取
1.3.1 PSSM 位置特异性得分矩阵特征
PSSM(Position Specific Scoring Matrix, PSSM) 位置特异性得分矩阵可以反映蛋白质序列中每个位置上不同碱基出现的频率,矩阵的行表示蛋白质序列的长度,矩阵的列表示构成蛋白质序列的20种残基,矩阵中的每个元素表示相应位置上碱基出现的频率。使用PSI-BLAST软件将未知序列与Swiss-Prot数据库中的序列进行多轮迭代的多序列比对,得到PSSM位置特异性得分矩阵,之后使用归一化函数sigmoid对矩阵中的数据进行归一化处理。归一化函数sigmoid计算公式如下:
其中x 代表矩阵中的原始数值,f(x)代表归一化后的数值。在应用大小为17的滑动窗口后,PSSM位置特异性得分矩阵特征的总维数为20×17=340。
1.3.2 蛋白质二级结构特征
蛋白质二级结构(Protein Secondary Structure) 是指多肽主链骨架原子沿一定的轴盘旋或折叠而形成的特定构象,即肽链主链骨架原子的空间位置排布,不涉及残基侧链。按照主流的分类方法,蛋白质的二级结构可分为三类,分别为:α-螺旋、β-折叠和无规卷曲。在本文中,使用PSIPRED工具预测蛋白质序列中每个残基属于α- 螺旋、β-折叠和无规卷曲三种结构的概率,得到三维的蛋白质二级结构特征数据。经过滑动窗口处理,该特征值总维数是3×17=51。
1.3.3 溶剂可及性特征
可及表面积(Acces?sible Surface Area, ASA) 或溶剂可及表面积(Solvent-Accessible Surface Area, SASA) 是溶剂可接触的生物分子表面积。蛋白质序列中的残基溶剂可及性表面积越大,越有可能与核苷酸发生反应。在本文中,使用ASAquick 工具预测蛋白质序列中每个残基的溶剂可及性表面积,预测结果以1位数值的形式给出。经过滑动窗口处理,该特征值总维数是1×17=17。
1.3.4 序列特征
根据蛋白质序列中氨基酸的偶极子和侧链数量,通过one-hot编码将20种氨基酸分别标识,Ala、Gly和Val用0000001表示,Ile、Leu、The和Pro用0000010表示,His、Asn、Gln和Trp用0000100表示,Tyr、Met、Thr 和Ser 用0001000 表示,Arg 和Lys 用0010000 表示,Asp和Glu用0100000表示,Cys用1000000表示,划分完后的氨基酸编码特征值共有7维[11]。经过滑动窗口处理,该特征值总维数是7×17=119。
1.3.5 残基的理化性质
本文共用了残基的支链, 分子量, 等电点, 羧基解离常数和氨基解离常数5种理化性质。对于以上五种理化性质,首先,根据残基的支链性质差异,可将20种残基分为疏水性残基、亲水性残基、碱性残基、酸性残基 。使用one-hot编码来对每个残基进行分类编码,该特征可用4维的one-hot值分别代表四种不同的残基类别,0001代表疏水性残基,0010代表亲水性残基,0100代表碱性残基,1000代表酸性残基。
其次,根据残基的分子量, 等电点, 羧基解离常数, 氨基解离常数可构成4维数据,每1维数据都采用最大最小值归一化方法进行归一化处理。最大最小值归一化函数如下:
其中x 代表残基的分子量(或等电点, 羧基解离常数, 氨基解离常数)的数值,min 代表残基的分子量(或等电点, 羧基解离常数, 氨基解离常数)数值的最小值, max 代表残基的分子量(或等电点, 羧基解离常数, 氨基解离常数)数值的最大值。经过滑动窗口处理,该特征值总维数是8×17=136。
1.3.6 标准输入数据处理
在对蛋白质序列进行不同方式的特征提取后,经过滑动窗口处理,得到了340维PSSM位置特异性得分矩阵特值、51维蛋白质二级结构特征值、17维溶剂可及性特征值、119维序列特征值和136 维残基理化性质特征值。所有特征值拼接后共663维,为了对应深度卷积网络的输入形式,对663×1的特征值进行数据变形处理,得到17×39×1的标准输入数据。
1.4 评价指标
本文研究的蛋白质-ATP绑定位点预测是正负样本不平衡的二分类问题,通过比较预测结果与真实标签的数值,计算特异性(Specificity) 、敏感性(Sensitiv?ity) 、准确性(Accuracy) 、马修斯相关系数(MCC) 和ROC曲线下与坐标轴围成的面积(AUC) 共五种评价指标来衡量本文提出的蛋白质-ATP绑定位点预测方法的整体性能。在二分类问题中,混淆矩阵可以比较直观地反映模型性能,混淆矩阵的计算方法如表1 所示。
本文使用的评价指标,其计算方法为:
AUC(Area Under Curve) 被定义为ROC曲线与坐标轴围成的面积。ROC曲线不固定阈值,可反映训练模型在全阈值下的情况。由于本文所涉及的是样本不平衡问题,AUC作为一种不依赖阈值的分类指标,不受到数据集正负样本比例的影响,能够更加客观全面地反映分类器的性能。
2 模型结构与训练
2.1 模型结构
本文采用InceptionV3 和SVM 相结合的模型结构,通过Inception V3网络结构对输入数据进行增维,SVM分类器对增维后的数据进行线性分类得到最终的预测结果,整体的模型结构如图2所示。
数据输入格式是17×39×1的张量,第一层是深度卷积层,由64个3×3卷积核、32个2×2卷积核、64个2×2卷积核、3×3的最大池化层、80个1×1卷积核、192 个2×2卷积核与3×3的最大池化层构成。第二层是InceptionV3 模块组,由256 个输出节点的InceptionModule 1、768 个输出节点的Inception Module 2 和2 048个输出节点的Inception Module 3构成,以上三种Inception Module的具体架构将在后续进行详细描述。第三层是数据平铺层,由3×3的平均池化层、保留率为0.5的Dropout层和通道数为1 000、输出节点为2的全连接层组成。第四层是SVM分类层,对平铺层处理后的数据进行二分类,根据数据集中正负样本数量比例计算得到正负样本权重分别为13.322 和0.519,正负样本将乘以相对应的权重,使得模型在训练过程中更加关注权重更高的正样本类,最后通过SVM分类器得出预测结果。
本文通过将InceptionV3和SVM两种分类算法进行结合,完成对蛋白质-ATP绑定位点的预测任务,下面将分别对两种分类算法进行简要介绍。
2.1.1 InceptionV3网络结构
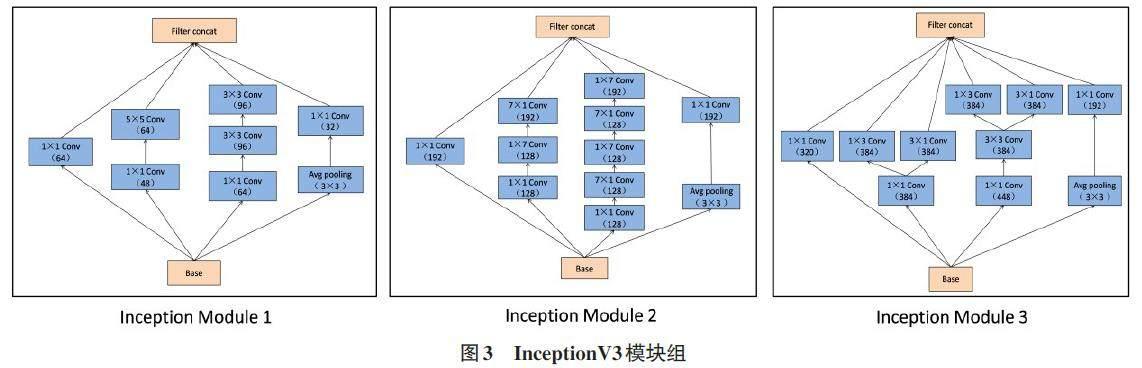
InceptionV3深度卷积网络通过把一个N×N的大卷积核拆分成多个1×N、N×1或者M×M(M 本文InceptionV3网络结构的详细参数如表2所示。Inception V3网络结构采用不同大小的卷积核提高数据识别能力,提取更加有效的局部特征,最后的数据拼接可以将不同尺度的特征进行融合,在改变不同网络层结构单一的基础上,提高了网络结构的计算能力。 其中InceptionV3 模块组中3 种Inception Module 的网络结构如图3 所示,从上到下依次是InceptionModule 1、Inception Module 2和 Inception Module 3。 2.1.2 支持向量机(Support Vector Machines, SVM) SVM是一类按监督学习(supervised learning) 方式对数据进行二元分类的广义线性分类器(generalizedlinear classifier) ,其决策边界是对学习样本求解的最大边距超平面(maximum-margin hyperplane) 。在蛋白质-ATP 绑定位点预测研究方面,已经有很多采用SVM分类器的方法[5-7],并取得了良好的预测结果,但在模型改进和方法融合方面仍有待提升。本文通过Inception V3网络结构对输入数据进行增维,将提取到的深层特征作为SVM分类器的输入,应用SVM优秀的分类性能,以达到更好的预测效果。使用的SVM 中详细参数如下所示: 对于边界点的惩罚系数C取值为0.8,C越大代表这个分类器对在边界内的噪声点的容忍度越小,分类准确率高,但是容易过拟合,泛化能力差。所以一般情况下,适当减小C,对在边界范围内的噪声有一定容忍。 由于使用的数据集不是线性可分的,需要利用核函数将数据集映射到高维空间,核函数采用高斯核函数,如公式(7) 所示: 其中x,y 是输入样本数据,γ 是超参数,一般取值为样本特征数的倒数,本文取值为0.5,|| x - y ||表示向量的范数,可以理解为向量的模,k 表示两个向量之间的关系,结果为一个具体的值。 启发式收缩方式shrinking设置为True,由于SVM 分类器接受的数据是经过Inception V3网络结构处理后的数据,能预知到哪些变量对应着支持向量,在不影响训练结果的基础上有助于迅速求解,起到一个加速训练的效果。停止训练的误差精度tol 设置为0.001。预测结果概率probability取值为True,使SVM 分类器以概率值的形式输出正负预测值,以便于最后的损失函数更加精确的计算损失值。 2.2 模型训练 2.2.1 样本不平衡处理 由于训练集正负样本的数量极不平衡,导致蛋白质-ATP绑定位点预测成为一类典型的不平衡学习问题。对于解决该问题的常用方式有上采样、下采样、设定类别权重(class_weight) 、设定样本权重(sample_weight) 等。上采样方法是通过向少数类的样本中增加新的样本的方式实现正负样本平衡,但是新的样本中可能存在冗余数据,影响训练准确性。下采样方法是从多数类样本中选取一些样本进行舍弃,这种方法的缺点是被舍弃的样本可能包含一些重要信息,致使学习出来的模型效果不好。sample_weight主要解决的是样本质量不同的问题,class_weight主要解决数据不平衡问题。因此本文采用设定类别权重的方法,通过设定class_weight参数来解决训练集正负样本不平衡问题,在充分利用所有数据的基础上,将不同的类别映射为不同的权值,该参数用来在训练过程中调整损失函数(只用于模型训练过程)。通过计算数据集中正负样本的数量比例,得出正负样本类别权重分别为13.322和0.519。该参数在处理非平衡的训练数据(正类的训练样本数很少)时,可以使得损失函数对样本数不足的数据更加关注,从而通过训练得到更精确的分类模型。 2.2.2 损失函数 正负样本不平衡带来的问题主要表现为样本中会存在大量的易分类样本,易分类负样本在分类器模型训练过程中对损失函数的loss值影响较大,网络结构从正样本数据中获取的信息较少,影响损失函数的梯度变化方向,无法对所有样本进行准确分类。由于本文研究的蛋白质-ATP绑定位点预测问题是二分类问题并且正负样本数量极不平衡,为了使模型在训练时更多地关注难分类正样本,可以使用Focal_ Loss(如公式(8)所示)与平衡交叉熵 balanced cross entropy(如公式(9)所示)相结合的损失函数,如公式(10)所示,其中p_t和α的定义如公式(11)和公式(12)所示。 其中p 是模型的预测值,y 是样本标签的真实值,α、γ 是超常,为常数,当α=1,γ=0时,FL 和普通交叉熵损失函数一致。α 权重因子的大小决定正负样本在损失函数中的比重,即负样本越多,给它的权重越小,这样就可以降低负样本的影响。 根据本文数据集中正负样本比例,α 取值为0.25,图4展示了当α=0.25时,γ 在不同的取值情况下,FL 的函数图像。本文通过多轮实验对比,当α=0.25,γ<2 时,损失函数的收敛速度较慢,模型学习效率较低;当α=0.25,γ>2时,损失函数的收敛速度较快,loss 值在迭代早期降得太快,训练后期容易过拟合;当α=0.25,γ=2时,损失函数的收敛效果和模型训练效果最佳。 3 实验结果与分析 3.1 InceptionV3 与RondomForest 和SVM 分别结合的分类架构性能对比 为了验证InceptionV3深度神经网络模型与不同机器学习算法相结合的分类架构在蛋白质-ATP绑定位点预测方面的训练泛化效果。本文对比了Incep?tionV3深度神经网络模型与支持向量机(SVM) 和随机森林(RondomForest) 分别结合后的分类架构在ATP-388数据集上的预测性能。采用5折交叉验证方法将ATP-388数据集分为训练集和验证集,训练100轮次结束,以验证集的AUC 值作为第一评估指标,选取AUC值最高的一组结果作为实验结果进行比较。表3 展示了两种不同分类架构在ATP-388验证集上的预测性能。 从表3中可以看出,InceptionV3_SVM分类架构在ATP-388 验证集上获得的AUC 值与MCC 值分别是0.886和0.629,比InceptionV3_RondomForest分类架构获得的AUC(0.885) 与MCC(0.627) 高出0.1% 和0.2%,表明InceptionV3_SVM分类架构在ATP-388验证集上的预测性能优于InceptionV3_RondomForest分类架构的预测性能,模型泛化能力更好。 3.2 InceptionV3_SVM 分类架构与不同卷积网络的性能对比 从表4 中,很容易发现本文提出的Incep?tionV3_SVM分类架构在ATP-388数据集上的预测性能明显优于普通卷积神经网络架构和InceptionV3深度卷积神经网络架构的预测性能。本文方法在ATP-388验证集上获得的AUC值与MCC值分别是0.887 和0.632,比普通卷积神经网络架构获得的AUC(0.816) 与MCC(0.615) 高出7.1% 和1.7%,比InceptionV3深度卷积神经网络架构获得的AUC(0.879) 与MCC(0.619) 高出0.8% 和1.3%。从比较结果上可以看出,本文提出的Incep?tionV3_SVM分类架构是一种泛化能力更好,预测精度更高的蛋白质-ATP绑定位点的分类架构。其性能提升的主要来源在于,传统卷积神经网络全连接层到输出层间通过数据加权求和的分类函数进行数据降维分类,而本文在全连接层和输出层之间加入SVM分类器,依靠SVM优秀的二分类能力,对InceptionV3网络增维后的数据构建最优分类超平面,提高分类器的预测性能,其分类能力要强于直接应用全连接层的降维方式。 3.3 本文方法与已有预测方法在ATP-41 独立测试集上的实验结果比较 由表5中数据可知,本文方法在独立测试集的预测结果为:AUC指标达到0.886,通过选取最佳阈值,Acc指标达到0.972,Spe指标达到0.993,Sen指标达到0.542,Mcc指标达到0.635。在与以往预测方法的性能比较中,本文提出的InceptionV3_SVM分类架构预测方法在主要评价指标AUC上超过了其他对比方法,Acc和Spe指标值也达到了较高水平。Sen和Mcc指标值略低于ATPseq方法,但与其他预测方法相比具有显著提升,其主要原因在于,ATPseq 方法使用SSITE和TM-SITE方法的输出作为一类特征,S-SITE 是一种使用基于模板的序列图谱检测蛋白质绑定位点的方法,TM-SITE是一种基于结构模板查询通用蛋白质绑定位点的方法。相比而言,本文方法的特征均来源于蛋白质原始序列信息,在预测方法易用性和可行性方面具有优势。实验结果表明本文提出的分类架构方法能够准确预测蛋白质-ATP绑定位点,且在预测性能上相比以往方法具有一定程度的提升。 3.4 独立测试集预测样例结果分析 为了验证InceptionV3_SVM 分类模型在蛋白质-ATP绑定位点预测方面的黑盒测试能力,从ATP-41 独立测试集中选取PDB_ID为3J8Y_K的蛋白质序列进行样例分析。通过对3J8Y_K蛋白质序列进行特征提取,用InceptionV3_SVM架构分类模型得出的预测结果如图5所示。其中折线代表InceptionV3_SVM分类模型预测出的绑定位点概率值连成的线,虚点直线代表进行类别判定时取值为0.3的阈值,第一行标记点代表3J8Y_K蛋白质序列中真实绑定位点的位置标签值,第二行标记点代表3J8Y_K蛋白质序列在Incep?tionV3_SVM分类模型处理后预测的绑定位点位置标签值。 3J8Y_K蛋白质序列330个氨基酸中,有15个绑定位点和315个非绑定位点。从图5中可以看出,通过InceptionV3_SVM架构分类模型预测出的结果中,有14个绑定位点,316个非绑定位点,其中真阳性结果有14个,假阳性结果有0个,真阴性结果有315个,假阴性结果有1个。通过评价指标公式进行计算,得出的Acc 指标为0.997,Spe 指标为1,Sen 指标为0.933,Mcc指标为0.965。从评价指标计算结果可以看出, InceptionV3_SVM分类模型在进行新蛋白质绑定位点预测时,其模型性能和预测结果都有一定的有效性,该模型表现出较优的数据识别能力和数据处理能力,可以实现对蛋白质数据库中未知蛋白质序列的ATP绑定位点进行预测。 4 讨论 本文主要研究了蛋白质-ATP 绑定位点预测问题,首先对蛋白质序列进行特征提取获得标准输入张量,通过设定class_weight参数解决训练集正负样本不平衡问题,然后基于InceptionV3卷积神经网络模型和SVM分类器提出InceptionV3_SVM分类架构,用以识别蛋白质-ATP绑定位点。实验结果表明本文提出的方法具有良好的预测性能,对于蛋白质-ATP绑定位点预测、蛋白质功能探索、生物医学和药物研发等相关领域的研究具有推动作用。 参考文献: [1] 杨荣武.分子生物学[M].2版.南京:南京大学出版社,2017. [2] BECK J,NASSAL M.Hepatitis B virus replication[J].World Jour?nal of Gastroenterology,2007,13(1):48-64. [3] SCAGGIANTE B,KAZEMI M,POZZATO G,et al.Novel hepato?cellular carcinoma molecules with prognostic and therapeutic potentials[J].World Journal of Gastroenterology,2014,20(5):1268-1288. [4] CHEN H B,GU Z J,AN H W,et al.Precise nano?medicine for intelligent therapy of cancer[J].Sci?ence China Chemistry,2018,61(12):1503-1552. [5] CHEN K,MIZIANTY M J,KURGAN L.ATPsite:sequence-based prediction of ATP-binding residues[J].Proteome Science,2011,9(Suppl 1):S4. [6] 石大宏.基于序列的蛋白质—核苷酸绑定位点预测研究[D].南京:南京理工大学,2015:22-34. [7] 余健浩,孙廷凯.基于随机下采样和SVR的蛋白质-ATP绑定位点预测[J].现代电子技术,2015,38(4):19-24. [8] SONG J Z,LIU G X,SONG C Y,et al.A novel sequence-based prediction method for ATP-binding sites using fusion of SMOTE algorithm and random forests classifier[J].Biotechnol?ogy & Biotechnological Equipment,2020,34(1):1336-1346. [9] 郭丽萍.基于深度学习的蛋白质-ATP绑定位点预测研究[D].长春:东北师范大学,2019:21-40. [10] 张寓,於东军.基于一维卷积神经网络的蛋白质-ATP绑定位点预测[J].计算机应用,2019,39(11):3146-3150. [11] 刘桂霞,裴志尧,宋佳智.基于深度学习的蛋白质-ATP结合位点预测[J].吉林大学学报(工学版),2022,52(1):187-194. [12] HU J,LI Y,ZHANG Y,et al.ATPbind:accurate protein-ATP binding site prediction by combining sequence-profiling and structure-based comparisons[J].Journal of Chemical Informa?tion and Modeling,2018,58(2):501-510. 【通联编辑:李雅琪】 基金项目:国家自然科学基金(项目编号:62162050) ;内蒙古民族大学博士科研启动基金项目(项目编号:KYQD23006) ;三亚学院中青年教师(科研类)培养项目(项目编号:USYJSPY24-52)
猜你喜欢
肝博士(2022年3期)2022-06-30 02:48:48
海外星云(2021年9期)2021-10-14 07:26:10
电子制作(2018年19期)2018-11-14 02:37:08
自动化学报(2017年11期)2017-04-04 02:52:58
青苹果·教育研究版(2016年9期)2016-12-23 11:52:36
噪声与振动控制(2015年4期)2015-01-01 07:08:21
轴承(2010年2期)2010-07-28 02:26:12
