
人工智能蛋白质设计技术的研究进展及在生物医药创新开发中的应用与面临的挑战
2024-05-20 15:16苗洪江董泽凯向秋茹薛贵荣
上海医药 2024年4期
关键词:人工智能
苗洪江 董泽凯 向秋茹 薛贵荣

摘 要 蛋白质是协调复杂生命过程的精密“分子机器”,具有巨大的医疗应用潜力。然而,因为蛋白质的一维氨基酸序列、三维结构和生物功能之间的关联复杂,所以设计蛋白质并将其工程化以实现预期的功能和特性是一个极其困难的挑战。目前,人工智能在各个领域均取得了革命性的进展,人工智能与蛋白质工程技术的结合已成为一种强大的新型蛋白质设计工具,可用于生成各类生物活性分子。本文介绍人工智能蛋白质模拟和设计领域的研究进展和应用,尤其是在生物医药创新开发应用中面临的挑战和前景。
关键词 人工智能 蛋白质工程 新型蛋白质 合成生物学 药物开发
中图分类号:O629.73; TP399 文献标志码:A 文章编号:1006-1533(2024)07-0001-09
引用本文 苗洪江, 董泽凯, 向秋茹, 等. 人工智能蛋白质设计技术的研究进展及在生物医药创新开发中的应用与面临的挑战[J]. 上海医药, 2024, 45(7): 1-9; 55.
Research progress of artificial intelligence powered protein design and the prospect and challenges of its application in innovative biologics design
MIAO Hongjiang, DONG Zekai, XIANG Qiuru, XUE Guirong
(Shanghai Tianrang Intelligence Co., Ltd., Shanghai 200232, China)
ABSTRACT Proteins, the intricate “molecular machines” that orchestrate lifes processes, hold immense potential for therapeutic applications. However, the designing and engineering of these proteins towards desired properties and functions remain a formidable challenge due to the complex interplay between the amino acid sequence, the three dimensional structure, and biological function. Artificial intelligence (AI) has been making transformative strides in various fields and its combination with protein engineering techniques offers a powerful toolkit in generating novel proteins for synthetic biology and therapeutics development. In this review, we will discuss the advancements and applications of AI in protein modeling and design and highlight the challenges and outlook of its applications.
KEY WORDS artificial intelligence; protein engineering; novel proteins; synthetic biology; therapeutics development
我国是全球第二大药品市场,但抗体药物市场仅占全球抗体药物市场份额的10%,在新型生物药物的研发和供给方面仍然面临着严峻挑战。近年来,随着人工智能(artificial intelligence, AI)技术的快速发展,AI在蛋白质工程领域也取得了突破性进展,不仅解决了困扰生物学领域超半个世纪的蛋白质结构预测难题[1],而且从头设计出了环肽[2-3]、抗体[4-6]、荧光素酶[7]、蛋白质开关[8]、自组装的蛋白质纳米颗粒[9]等各种类型的全新功能蛋白,为生物医药、合成生物学等领域带来了大量全新的具有成药潜力的生物活性分子。本文介绍AI在蛋白质模拟和设计领域的研究进展,以及相关研究进展在生物医药创新开发中的应用情况和潜在挑战,为相关研发和从业人员展现一幅清晰的AI蛋白质设计技术发展图谱,以启发对蛋白质设计领域中新技术的探索开发和实践应用,共同解决人们尚未得到满足的医疗需求。
1 蛋白质结构预测
蛋白质参与生命过程的各个方面,如细胞信号转导、基因修正和复制、新陈代谢调控等,是人体中最重要的分子类型之一。蛋白质的生物功能与其三维结构密切相关,蛋白质结构的测定一直是生物制药、合成生物学等众多领域研究的核心基础:一方面,以X线晶体衍射、冷冻电镜为代表的蛋白质结构实验测定方法技术门槛高、耗时长,难以满足生命科学研究对蛋白质结构解析的通量需求;另一方面,通过计算预测蛋白质结构、解释蛋白质折叠的原理被视为分子生物学研究的“圣杯”,但由于蛋白质的生物复杂度高、潜在三维结构空间巨大,蛋白质的準确结构预测难题困扰了计算生物学领域长达半个多世纪。本章节主要介绍AI在蛋白质结构预测领域中的研究进展和其中部分具有代表性的方法及其应用。
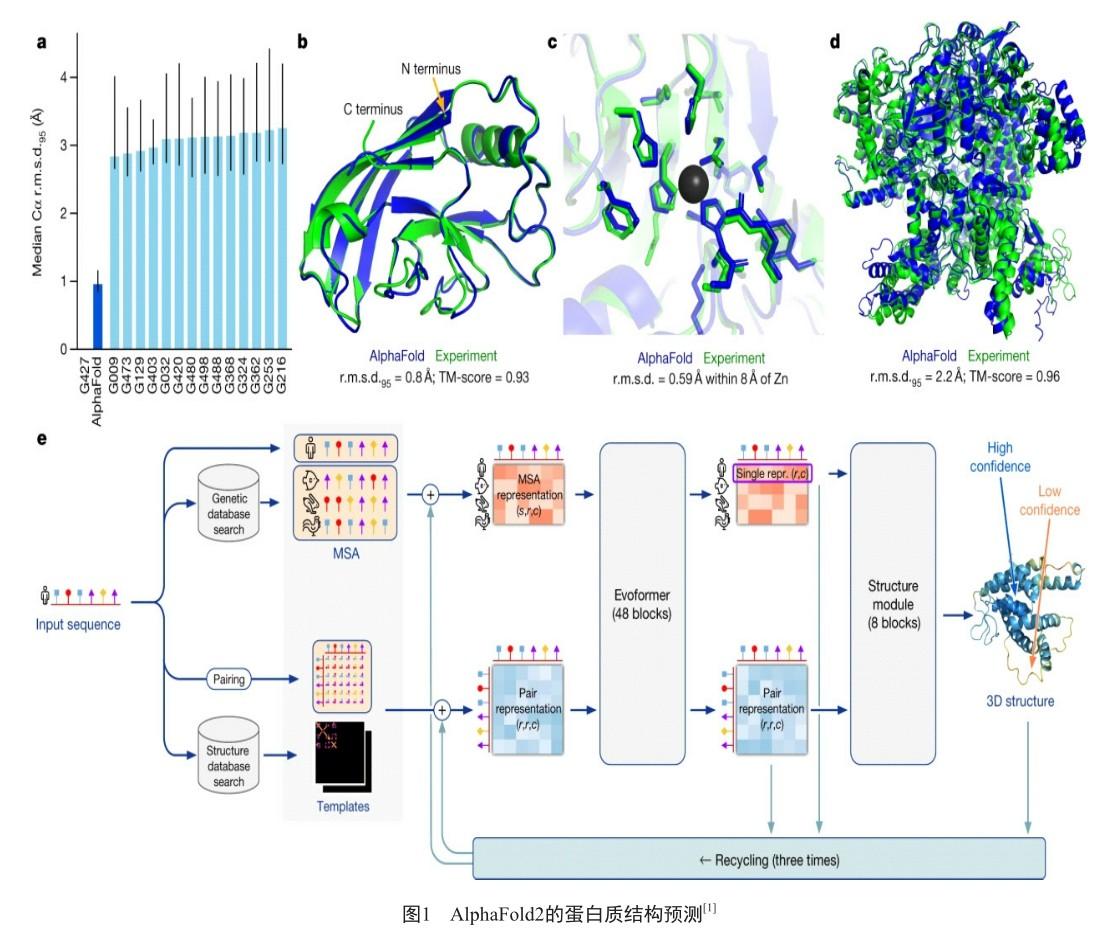
1.1 AI破解蛋白质结构预测难题
传统的基于物理力场或知识能量函数的蛋白质三维结构预测方法,如ROSETTA[10]、TINKER[11]等,不仅需要进行大量的计算以寻找能量最小化的蛋白质三维构象,而且预测的精度往往也较低。另有不少研发人员开发了各种同源建模方法[12-15],根据序列同源性从已知蛋白质结构数据库中寻找高度相似的结构模板来完成预测,虽然计算效率显著提高,但由于算法依赖并受限于高质量结构模板,适用范围十分有限。Wang等[16]首次将残差神经网络用于蛋白质残基间的接触图矩阵预测,再将接触图矩阵作为限制条件进行从头模拟,使可预测的蛋白质数量提高了近3倍。谷歌DeepMind团队开发的AlphaFold2[1]采用transformer架构从多序列比对(multiple sequence alignment, MSA)中抽取氨基酸残基间的关联信息,再通过具备旋转平移不变性的结构模块映射到三维空间,端到端的实现了近原子级别精确度的蛋白质结构预测,标志着蛋白质结构预测难题已获破解(图1)。DeepMind团队运用此算法开展了大规模的蛋白质结构预测工作,并与欧洲分子生物学实验室和欧洲生物信息研究所合作建立和开放了蛋白质结构数据库,数据库中的蛋白质三维结构至2023年6月已突破2亿个,涵盖了已知的各种生物来源的蛋白质[17]。DeepMind团队还于近期推出了更新版本的AlphaFold-latest[18],后者可同时对蛋白质及其各类配体,如小分子、核酸、金属离子、被修饰的残基等形成的复合体结构进行预测,且预测准确度也大大超越之前的SOTA模型。
与此同时,国内外研发人员还开发了RoseTTAFold[19]、TRFold2[20]、Uni-Fold[21],OpenFold[22]等一系列蛋白质结构预测准确度与AlphaFold2相当的算法。此外,Wu等[23]和Lin等[24]分别提出了OmegaFold和ESMFold算法,基于经大量蛋白质序列预训练获得的蛋白质语言模型,直接从目标蛋白质序列中提取高维嵌入信息,再通过结构模块映射到三维空间,避免了对MSA的依赖,对孤儿蛋白质(orphan protein)、抗体等类型蛋白质的结构预测准确度更高。
1.2 AI预测蛋白质结构的广泛应用
隨着AlphaFold2等各类能够准确预测蛋白质结构的AI模型的开源和大规模蛋白质结构数据库的开放,AI预测的蛋白质结构被广泛应用于生命科学研究、生物医药开发和合成生物学探索等各个领域。
Huang等[25]运用AlphaFold2预测了脱氨酶家族中所有蛋白质的三维结构,并根据结构相似性聚类发现了多个新型脱氨酶,这些脱氨酶经简单的工程化改造后可由单个腺病毒载体递送,首次实现了大豆植物内的胞嘧啶碱基编辑。Kreitz等[26]运用AlphaFold2对Photorhabdus virulence cassette的尾纤维蛋白进行模拟和工程化改造,开发出新型蛋白质靶向递送系统,为基因编辑、癌症治疗和生物调控提供了新的途径。Ren等[27]基于AlphaFold2预测的结构生成了细胞周期蛋白依赖性激酶20的高抑制性小分子,优化后的小分子已进入临床试验开发阶段。Ko等[28]结合运用冷冻电镜解析与AlphaFold2预测获得了配子表面蛋白质Pfs48/45的准确结构,进而确定了疟疾传播阻断抗体的结合位置并从中选出了合适片段进行疟疾疫苗的开发。
2 蛋白质设计
依赖自然进化获得新的蛋白质功能和特性通常需要数百年乃至数万年的时间,这难以满足当前人类在疾病检测和治疗、工业合成和生产等众多领域的蛋白质需求。此外,蛋白质还存在巨大的未被探索空间,以一种由100个氨基酸组成的蛋白质为例,其可能的序列有2100之多,远远超出人类已知的天然蛋白质序列的总和。随着基因检测及其编辑技术的快速发展,科学家们已可高效地合成指定蛋白质的基因序列,从而通过各类细胞或无细胞表达方法获得这些蛋白质[29-30]。然而,要从如此庞大的蛋白质序列空间中获得具备预期功能和特性的蛋白质子集是非常困难的。本章节主要介绍AI在蛋白质定向进化和从头设计领域中的研究进展与应用。
2.1 定向进化蛋白质序列优化
定向进化是一种通过模拟自然进化过程对天然蛋白质进行的有目的的改造方法[31],其先以饱和突变或随机突变的方式建立包含大量突变体的文库,然后对文库中的序列进行实验验证以获得具备预期功能或特性的优化突变体,之后再组合上一轮被验证的优化突变位点及氨基酸类型来构建新的突变体文库,通过多轮实验,最终获得功能更强、特性更优并可满足应用需求的蛋白质序列。然而,鉴于大多数随机突变是有害突变的现实,定向进化常需通过高通量、多轮次的实验来完成,投入高、周期长,且因只能对有限突变点位进行探索,结果可能落入局部最优解陷阱而导致实验失败。
AI技术的应用大幅改善了定向进化的效率和成功率。得益于AI算法,研发人员能在计算机上对庞大的蛋白质适应度空间进行全面探索,避免落入局部最优解陷阱。同时,AI模型可通过学习大量的特性标记的生物实验数据或采用无监督的方式经学习千万级的氨基酸序列来获取序列与特性的关联,故能进行更加精准的突变体文库建设,减少实验验证的量级和轮次,大幅降低优化突变体筛选成本,使定向进化具备更好的时效性和产业应用价值。Biswas等[32]开发了基于预训练模型的low-N设计法,仅依赖24条有标签数据训练的下游模型指导定向进化,基于同一野生型维多利亚绿色荧光蛋白酶,只通过1轮实验便获得了活性超过文献报告的经多轮定向进化所得优化突变体的新序列。Kulikova等[33]开发的MutCompute采用三维卷积神经网络学习蛋白质结构的局部适应度空间,用于预测具备潜在增益效果的氨基酸突变。运用此方法,Lu等[34]开发出与天然聚对苯二甲酸乙二醇酯(polyethylene terephthalate, PET)水解酶相比有5个点位突变的FAST-PETase,后者在各种温度和pH环境下的活性均更好,仅需1周时间便可完全降解来自51种热成型产品的未经任何预处理的PET。Hie等[35]运用完全无监督的蛋白质语言模型推荐进化性可行的突变,结果仅经2轮和总数不超过20个的实验筛选便获得了亲和力高于临床开发阶段抗体的突变体。
2.2 蛋白质从头设计
蛋白质从头设计不再依赖已知的天然蛋白质,而是根据预期功能或结构直接设计蛋白质的氨基酸序列,以实现从无到有的具有全新功能的蛋白质开发[36]。这种从零开始的设计虽然具有广泛的应用价值和广阔的应用前景,但由于目前对蛋白质序列-结构-功能深层关联的认知尚很有限,蛋白质从头设计是一项极具挑战性的工作。得益于前沿生成式AI技术的快速发展,蛋白质从头设计,无论是基于结构还是基于序列的方法,都取得了突破性的进展[37-39]。
2.2.1 基于结构的蛋白质从头设计
由于蛋白质的氨基酸序列决定蛋白质的三维结构,三维结构又决定了蛋白质的功能,故基于结构的蛋白质设计方法多以结构作为支点,先找到符合预期功能的蛋白质结构,再挖掘可折叠成此结构的氨基酸序列。Huang等[40]开发了SCUBA,采用神经网络模拟以主链为中心且独立于侧链的能量函数,先用基于卷积核密度估计的方法从蛋白质结构中估算统计能量函数,然后训练全连接的神经感知机来表达此函数。进行蛋白质设计任务时,通过SCUBA驱动的随机动力学模拟生成可设计的主链,再运用研发人员自主开发的ABACUS2[41]为主链填充氨基酸序列。Huang等[40]在实验室中合成了由SCUBA设计的蛋白质并通过X线晶体衍射解析了其结构,验证发现设计与实际结构间的误差为0.96~1.85 ?,达到了原子级别精度。
得益于蛋白质结构预测难题的破解,不仅蛋白质设计结果可借助预测模型来进行高效、准确的计算检验,而且能直接将预测模型应用于蛋白质设计流程以同时生成序列和结构,进一步提高蛋白质设计的效率和成功率。Wang等[42]开发的RFDesign、天壤XLab[20]开发的TRDesign等都是以结构预测模型为底座的蛋白质从头设计方法:通过设置与目标相关的模体损失函数、幻想损失函数等,以马尔科夫链蒙特卡洛(Markov chain Monte Carlo)或梯度下降的方式对初始序列进行突变优化,直到完成预设的优化轮次或获得符合预期的结构和序列。Wang等[42]还提出了蛋白质补齐(protein inpainting)的理念,与自然语言完形填空或从毁坏图像中恢复信息类似,在训练过程中掩盖局部的序列或结构使模型具备补齐能力。运用此方法设计的程序性细胞死亡受体配体-1结合蛋白pdl1_inp_1经实验证实不仅具有结合能力,且亲和力(Kd=326 nmol/L)也高于野生型的程序性细胞死亡受体-1(Kd=3.9 mmol/L),而其与已知蛋白质库中最接近的序列相似度只有25.4%。
去噪扩散模型[43]通过在前向过程中反复向数据中注入高斯噪声,再在反向过程中训练神经网络将高斯噪声去噪使模型获得生成能力。去噪扩散模型在图像和文本生成中有极好表现[44],用于蛋白质设计能进一步提高设计的结构新颖性、可控性和通用性。Trippe等[45]开发的ProtDiff采用具备不变性的图神经网络模拟和实现蛋白质主链的生成,并提出了一种顺序蒙特卡洛(sequential Monte Carlo)的估算方法SMCDiff来实现目标模体的设计,可在分钟级时间内完成设计工作。Watson等[46]的RFdiffusion在蛋白质折叠模型RoseTTAFold的基础上进行了基于结构去噪的微调,通过在主链坐标中加入三维高斯噪声和模拟布朗运动在蛋白质的结构中加入平移、旋转噪声,再在反向降噪中训练模型最小化预测和真实结构的均方差,设计时便能从随机初始化的结构中通过去噪生成全新的蛋白质主链结构了。Watson等[46]运用RFdiffusion生成了拓扑限制的单链蛋白质、蛋白质结合配体、对称性复合体、金属结合的蛋白质等各类全新的蛋白质并进行了系列生物实验验证,仅合成不足100条从头设计的序列就获得了甲型流感病毒血凝素H1、白介素-7受体α、程序性细胞死亡受体配体-1和原肌球蛋白受体激酶A的高亲和力结合配体。Generate:Biomedicines公司开发的Chroma系统[47]采用外部条件限制下的贝叶斯采样方法生成全新的蛋白质结构和序列,只需根据目标设置相应的限制条件,如对称性、局部构象、形状甚至自然语言提示,便可完成各类蛋白质的设计任务而不需要重新训练(图2)。
虽然许多模型能够同时进行蛋白质结构和序列的设计,但由于表面疏水性残基等原因会出现溶解性差、合成成功率低的问题[48],故常需对氨基酸序列进行进一步的优化。Dauparas等[49]提出了为固定蛋白质主链填补氨基酸序列的图神经网络ProteinMPNN,该模型采用编码器-解码器的结构从输入的主链结构计算图中提取节点和边的特征,采用循环的方式根据节点特征和已解码序列依次计算下一个残基点位的氨基酸分布概率并进行采样,实现了各类复杂结构的序列设计。经多项生物实验验证,ProteinMPNN可以很高的成功率获得较传统方法设计的序列和天然序列合成特性更优的氨基酸序列[50-52]。Hsu等[53]开发了ESM-IF,采用带有几何向量感知机层的图神经网络在一个由近20万个实验解析蛋白质结构和1 200万个由AlphaFold2预测的蛋白质结构组成的数据库上进行训练,在新冠病毒的刺突蛋白受体结合区域实现了50%以上的序列恢复率。
2.2.2 基于序列的蛋白质从头设计
与基于结构的蛋白质设计方法不同,基于序列的蛋白质设计方法希望根据功能或特性需求直接生成相应的氨基酸序列,生成对抗网络(generative adversarial network, GAN)和变分子编码器(variantional autoencoder, VAE)等生成式算法被广泛应用于此领域。Hawkins-Hooker等[54]开发了MSA-VAE和AR-VAE,通过编码器-解码器架构学习近7万条各种荧光素酶序列,设计生成的一些序列在生物检测中显示有发光效果。Repecka等[55]开发的ProteinGAN采用时间卷积网络同时学习序列中的局部和全局信息,由鉴别器网络对生成器网络产生的序列进行打分,由此不断提高模型生成符合天然蛋白质特性的序列的能力。在以细菌苹果酸脱氢酶为对象的生物实验中,一些由ProteinGAN生成的序列被证实具有苹果酸脱氢酶活性且有较好的溶解性。
随着自然语言模型的爆发式发展,针对蛋白质序列的蛋白质语言模型展现出了对蛋白质序列空间的极强的探索能力,即使不进行有监督的微调,也能仅基于蛋白质的序列信息零样本的学习对其功能及溶解性、热稳定性、结合位点等进行有效的预测[56-58]。基于这类预训练蛋白质语言模型,Saleforce Research公司等开发了ProGen[59],在以2.8亿条蛋白质序列及其相应功能和特性作为限制标签的数据集上,将序列生成作为下一标记预测任务进行训练,而在指定的蛋白质家族上的微调可进一步提高ProGen的序列生成能力。设计时,ProGen可以根据输入的限制标签从头生成氨基酸序列。在以溶菌酶为对象的设计案例中,一些由ProGen生成的序列在体外实验中被证实具有催化活性,其中包括与天然蛋清溶菌酶活性相当的个体。Evozyne和英伟达公司联合开发了ProT-VAE[60],在蛋白質语言模型ProtT5的编码器和解码器中间通过卷积压缩和解压层链接嵌入可训练的VAE模型,在蛋白质设计时冻结蛋白质语言模型和中间层参数,仅训练中间的VAE就实现了高效的模型微调。由ProT-VAE设计的蛋白质经生物实验验证,生物活性较天然人苯丙氨酸羟化酶高2.5倍。
擴散模型亦被应用于基于序列的蛋白质从头设计。微软公司AI4Science团队等开发的EvoDiff[61]采用离散扩散实现蛋白质序列的从头设计,不仅能生成基于结构的设计方法无能为力的内在无序的蛋白质,而且模型可从用户输入的MSA中获取进化信息并指导生成,使设计的序列更好地拟合预期功能和特性。基因泰克公司Prescient团队提出了扩散优化采样在抗体序列上进行梯度指导的离散扩散,融合贝叶斯优化进行多目标指导的设计算法[62]。体外实验显示,应用此方法生成的针对人表皮生长因子2的抗体实现了97%的表达率和25%的结合率。
3 小结与展望
AI技术的应用已使蛋白质结构预测和设计领域获得了革命性的突破,不仅破解了困扰生物学家半个多世纪的蛋白质结构预测难题,而且各种由AI设计的全新蛋白质也在实验室中被合成出和得到验证,在生物医药、合成生物学等产业的应用正在不断展开。
美国蛋白质设计研究院与韩国SK生物科技公司合作开发的新冠病毒疫苗SKYCovione[63]已获准在韩国和英国上市并进行了大规模的接种。这种疫苗通过人工设计的自组装纳米蛋白质颗粒实现抗原表位的多次展示,引发的免疫反应和抗体激活效果显著高于传统疫苗,且因设计的抗原蛋白质具有很好的稳定性,疫苗无需冷冻保存和运输。Absci公司基于零样本生成式AI算法设计的抗人表皮生长因子2抗体不仅在表面等离子共振实验检测中显示有较曲妥珠单抗更强的亲和力,在免疫原性和各项可开发性指标上也均表现优异[64]。Absci公司已与默沙东、阿斯利康等公司达成合作协议,共同推进肿瘤、皮肤病等疾病治疗药物的开发。基于Chroma蛋白质从头设计平台,Generate:Biomedicines公司正在开发肿瘤、传染性疾病和免疫性疾病等治疗药物,并与Roswell Park综合癌症中心合作开发新型嵌合抗原受体T细胞疗法产品[65]。
未来,高通量的蛋白质生产和检验方法的开发和进步将持续扩大蛋白质设计的应用场景。例如,与传统的合成方法相比较,逐渐成熟的无细胞蛋白质合成技术具有更强的可控性、更低的经济和时间成本、更高的安全性[66-67],这会大幅加速设计—合成—检验—分析优化的流程,使蛋白质设计项目得以快速推进。此外,AI模型的训练需要大量精准标记的高质量数据,但在分子生物学领域,这样的数据是昂贵和稀缺的,是目前限制蛋白质设计落地应用的主要“瓶颈”,而全新的高通量检测方法恰能很好地应对此难题。A-Alpha Bio公司开发出独特的噬菌体展示技术,能同时展示数百万到数十亿个噬菌体,在保证灵敏度和特异度的前提下将蛋白质-配体结合的检测通量提高了几个数量级[68]。Porebski等[69]基于Illumina HiSeq平台开发的深度筛查技术可在3 d内完成数千万的抗体-抗原结合检测。进一步的,机器人和自动化实验室的部署还能对蛋白质的序列、特性和适应度空间进行更加全面的探索,分布更加均匀的数据有望继续提高AI设计方法的普适性[70-72]。
得益于广泛的关注和深入的研究,AI领域的最前沿模型正被迅速地应用于蛋白质设计,本文列举的设计方法的进化和设计能力的提高正是这种趋势的完美体现。虽然深度学习模型经常因为“黑盒”属性被诟病,但其在复杂生物问题的解释,如蛋白质结构预测、突变分析、蛋白质设计等领域,已经展现出远超基于专家经验或严谨的数学建模的水平[73]。研发人员也正通过在数据处理、模型架构设计、损失函数设置等各个环节嵌入生物物理和生物化学方面的经验和知识,提高AI模型的可解释性和鲁棒性[74-76]。
AI技术的广泛应用无疑加快了数字生物学时代到来的步伐,在算法快速发展、模型快速迭代更新的时代背景下,确保最前沿AI模型的可及性和民主化有助于提高整个蛋白质设计领域的产学研结合水平,促进“人工智能+生物技术”的产业化落地。因此,众多研发人员和公司都选择拥抱开源,让更多的研发和从业人员可以运用最先进和最符合需求的AI算法来解决目标问题。然而,当前的蛋白质设计方法普遍具有算法复杂、算力需求大、流程整合困难的特点,单纯的算法开源很难满足非计算背景研发人员的应用需求。为此,Cradle Bio公司、InstaDeep公司、天壤XLab等都推出了AI赋能的计算平台(图3),不仅配备充足的算力支持,而且对众多的蛋白质设计工具进行优化和整理,通过逻辑清晰、操作简单的页面将丰富的蛋白质模拟和设计能力展现给用户,使AI模型更加易用好用。
随着AI蛋白质设计技术的普及和应用,蛋白质设计将会越来越广泛地应用于多肽、抗体、酶等各类生物医药相关蛋白质的开发,从而推动蛋白质科学向蛋白质工程的转变。可以预见,按需设计的定制化、个体化精准医疗的出现已不再遥远。
参考文献
[1] Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold [J]. Nature, 2021, 596(7873): 583-589.
[2] Rettie SA, Campbell KV, Bera AK, et al. Cyclic peptide structure prediction and design using AlphaFold [EB/OL].[2023-11-15]. https://doi.org/10.1101/2023.02.25.529956.
[3] Kosugi T, Ohue M. Design of cyclic peptides targeting protein-protein interactions using AlphaFold [J]. Int J Mol Sci, 2023, 24(17): 13257.
[4] Pooja Mahajan S, Ruffolo J, Frick R, et al. Towards deep learning models for target-specific antibody design [J]. Biophys J, 2022, 121(3): 528a.
[5] Chungyoun M, Gray JJ. AI models for protein design are driving antibody engineering [J]. Curr Opin Biomed Eng, 2023, 28: 100473.
[6] Makowski EK, Chen HT, Tessier PM. Simplifying complex antibody engineering using machine learning [J]. Cell Syst, 2023, 14(8): 667-675.
[7] Yeh AH, Norn C, Kipnis Y, et al. De novo design of luciferases using deep learning [J]. Nature, 2023, 614(7949): 774-780.
[8] Pillai A, Idris A, Philomin A, et al. De novo design of allosterically switchable protein assemblies [EB/OL]. [2023-11-15]. https://doi.org/10.1101/2023.11.01.565167.
[9] Wicky BIM, Milles LF, Courbet A, et al. Hallucinating symmetric protein assemblies [J]. Science, 2022, 378(6615):56-61.
[10] Leaver-Fay A, Tyka M, Lewis SM, et al. ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules [J]. Methods Enzymol, 2011, 487: 545-574.
[11] Rackers JA, Wang Z, Lu C, et al. Tinker 8: software tools for molecular design [J]. J Chem Theory Comput, 2018, 14(10): 5273-5298.
[12] Yang J, Yan R, Roy A, et al. The I-TASSER Suite: protein structure and function prediction [J]. Nat Methods, 2015, 12(1): 7-8.
[13] Kelley LA, Sternberg MJ. Protein structure prediction on the web: a case study using the Phyre server [J]. Nat Protoc, 2009, 4(3): 363-371.
[14] Kelley LA, Mezulis S, Yates CM, et al. The Phyre2 web portal for protein modeling, prediction and analysis [J]. Nat Protoc, 2015, 10(6): 845-858.
[15] Waterhouse A, Bertoni M, Bienert S, et al. SWISS-MODEL: homology modelling of protein structures and complexes [J]. Nucleic Acids Res, 2018, 46(W1): W296-W303.
[16] Wang S, Sun S, Li Z, et al. Accurate de novo prediction of protein contact map by ultra-deep learning model [J]. PLoS Comput Biol, 2017, 13(1): e1005324.
[17] Tunyasuvunakool K, Adler J, Wu Z, et al. Highly accurate protein structure prediction for the human proteome [J]. Nature, 2021, 596(7873): 590-596.
[18] Google DeepMind AlphaFold Team, Isomorphic Labs Team. Performance and structural coverage of the latest, in-development AlphaFold model [EB/OL]. [2023-11-15]. https://storage.googleapis.com/deepmind-media/DeepMind. com/Blog/a-glimpse-of-the-next-generation-of-alphafold/ alphafold_latest_oct2023.pdf.
[19] Baek M, DiMaio F, Anishchenko I, et al. Accurate prediction of protein structures and interactions using a three-track neural network [J]. Science, 2021, 373(6557): 871-876.
[20] 上海天壤智能科技有限公司. 天壤XLAB [EB/OL]. [2023-11-15]. https://xlab.tianrang.com/xlab.
[21] Li Z, Liu X, Chen W, et al. Uni-Fold: an opensource platform for developing protein folding models beyond AlphaFold [EB/OL]. [2023-11-15]. https://doi. org/10.1101/2022.08.04.502811.
[22] Ahdritz G, Bouatta N, Floristean C, et al. OpenFold: retraining AlphaFold2 yields new insights into its learning mechanisms and capacity for generalization [EB/OL]. [2023-11-15]. https://doi.org/10.1101/2022.11.20.517210.
[23] Wu R, Ding F, Wang R, et al. High-resolution de novo structure prediction from primary sequence [EB/OL]. [2023-11-15]. https://doi.org/10.1101/2022.07.21.500999.
[24] Lin Z, Akin H, Rao R, et al. Evolutionary-scale prediction of atomic-level protein structure with a language model [J]. Science, 2023, 379(6637): 1123-1130.
[25] Huang J, Lin Q, Fei H, et al. Discovery of deaminase functions by structure-based protein clustering [J]. Cell, 2023, 186(15): 3182-3195.
[26] Kreitz J, Friedrich MJ, Guru A, et al. Programmable protein delivery with a bacterial contractile injection system [J]. Nature, 2023, 616(7956): 357-364.
[27] Ren F, Ding X, Zheng M, et al. AlphaFold accelerates artificial intelligence powered drug discovery: efficient discovery of a novel CDK20 small molecule inhibitor [J]. Chem Sci, 2023, 14(6): 1443-1452.
[28] Ko KT, Lennartz F, Mekhaiel D, et al. Structure of the malaria vaccine candidate Pfs48/45 and its recognition by transmission blocking antibodies [J]. Nat Commun, 2022, 13(1): 5603.
[29] Hoose A, Vellacott R, Storch M, et al. DNA synthesis technologies to close the gene writing gap [J]. Nat Rev Chem, 2023, 7(3): 144-161. Erratum in: Nat Rev Chem, 2023, 7(8): 590.
[30] Garenne D, Haines MC, Romantseva EF, et al. Cell-free gene expression [J]. Nat Rev Methods Primers, 2021, 1(1): 49.
[31] Xiong W, Liu B, Shen Y, et al. Protein engineering design from directed evolution to de novo synthesis [J]. Biochem Eng J, 2021, 174: 108096.
[32] Biswas S, Khimulya G, Alley EC, et al. Low-N protein engineering with data-efficient deep learning [J]. Nat Methods, 2021, 18(4): 389-396.
[33] Kulikova AV, Diaz DJ, Loy JM, et al. Learning the local landscape of protein structures with convolutional neural networks [J]. J Bio Phys, 2021, 47(4): 435-454.
[34] Lu H, Diaz DJ, Czarnecki NJ, et al. Machine learning-aided engineering of hydrolases for PET depolymerization [J]. Nature, 2022, 604(7907): 662-667.
[35] Hie BL, Shanker VR, Xu D, et al. Efficient evolution of human antibodies from general protein language models [J/ OL]. Nat Biotechnol, 2023 Apr 24. [2023-11-15]. https://doi. org/10.1038/s41587-023-01763-2.
[36] Woolfson DN. A brief history of de novo protein design: minimal, rational, and computational [J]. J Mol Biol, 2021, 433(20): 167160.
[37] Pan X, Kortemme T. Recent advances in de novo protein design: principles, methods, and applications [J]. J Biol Chem, 2021, 296: 100558.
[38] Ferruz N, Heinzinger M, Akdel M, et al. From sequence to function through structure: deep learning for protein design [J].Comput Struct Biotechnol J, 2022, 21: 238-250.
[39] Khakzad H, Igashov I, Schneuing A, et al. A new age in protein design empowered by deep learning [J]. Cell Syst, 2023, 14(11): 925-939.
[40] Huang B, Xu Y, Hu X, et al. A backbone-centred energy function of neural networks for protein design [J]. Nature, 2022, 602(7897): 523-528.
[41] Xiong P, Hu X, Huang B, et al. Increasing the efficiency and accuracy of the ABACUS protein sequence design method [J]. Bioinformatics, 2020, 36(1): 136-144.
[42] Wang J, Lisanza S, Juergens D, et al. Scaffolding protein functional sites using deep learning [J]. Science, 2022, 377(6604): 387-394.
[43] Ho J, Jain A, Abbeel P. Denoising diffusion probabilistic models [J]. Adv Neural Inf Process Syst, 2020, 33: 6840-6851.
[44] Lugmayr A, Danelljan M, Romero A, et al. Repaint: inpainting using denoising diffusion probabilistic models [EB/OL].[2023-11-15]. https://doi.org/10.48550/arXiv.2201.09865.
[45] Trippe BL, Yim J, Tischer D, et al. Diffusion probabilistic modeling of protein backbones in 3D for the motif-scaffolding problem [EB/OL]. [2023-11-15]. https://doi.org/10.48550/ arXiv.2206.04119.
[46] Watson JL, Juergens D, Bennett NR, et al. De novo design of protein structure and function with RFdiffusion [J]. Nature, 2023, 620(7976): 1089-1100.
[47] Ingraham JB, Baranov M, Costello Z, et al. Illuminating protein space with a programmable generative model [J]. Nature, 2023, 623(7989): 1070-1078.
[48] Goverde CA, Wolf B, Khakzad H, et al. De novo protein design by inversion of the AlphaFold structure prediction network [J]. Protein Sci, 2023, 32(6): e4653.
[49] Dauparas J, Anishchenko I, Bennett N, et al. Robust deep learning-based protein sequence design using ProteinMPNN[J]. Science, 2022, 378(6615): 49-56.
[50] Nikolaev A, Kuzmin A, Markeeva E, et al. Reengineering of a flavin-binding fluorescent protein using ProteinMPNN [EB/OL]. [2023-11-15]. https://doi. org/10.1101/2023.08.25.554855.
[51] Kao HW, Lu WL, Ho MR, et al. Robust design of effective allosteric activators for Rsp5 E3 ligase using the machine learning tool ProteinMPNN [J]. ACS Synth Biol, 2023, 12(8): 2310-2319.
[52] Sumida KH, Nú?ez-Franco R, Kalvet I, et al. Improving protein expression, stability, and function with ProteinMPNN [EB/OL]. [2023-11-15]. https://doi. org/10.1101/2023.10.03.560713.
[53] Hsu C, Verkuil R, Liu J, et al. Learning inverse folding from millions of predicted structures [EB/OL]. [2023-11-15]. https://doi.org/10.1101/2022.04.10.487779.
[54] Hawkins-Hooker A, Depardieu F, Baur S, et al. Generating functional protein variants with variational autoencoders [J]. PLoS Comput Biol, 2021, 17(2): e1008736.
[55] Repecka D, Jauniskis V, Karpus L, et al. Expanding functional protein sequence spaces using generative adversarial networks[J]. Nat Mach Intell, 2021, 3(4): 324-333.
[56] Alley EC, Khimulya G, Biswas S, et al. Unified rational protein engineering with sequence-based deep representation learning [J]. Nat Methods, 2019, 16(12): 1315-1322.
[57] Rives A, Meier J, Sercu T, et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences [J]. Proc Natl Acad Sci U S A, 2021, 118(15): e2016239118.
[58] Elnaggar A, Heinzinger M, Dallago C, et al. ProtTrans: toward understanding the language of life through selfsupervised learning [J]. IEEE Trans Pattern Anal Mach Intell, 2022, 44(10): 7112-7127.
[59] Madani A, Krause B, Greene ER, et al. Large language models generate functional protein sequences across diverse families [J]. Nat Biotechnol, 2023, 41(8): 1099-1106.
[60] Sevgen E, Moller J, Lange A, et al. ProT-VAE: Protein Transformer Variational AutoEncoder for functional protein design [EB/OL]. [2023-11-15]. https://doi. org/10.1101/2023.01.23.525232.
[61] Alamdari S, Thakkar N, van den Berg R, et al. Protein generation with evolutionary diffusion: sequence is all you need [EB/OL]. [2023-11-15]. https://doi. org/10.1101/2023.09.11.556673.
[62] Gruver N, Stanton S, Frey N, et al. Protein design with guided discrete diffusion [EB/OL]. [2023-11-15]. https://doi. org/10.48550/arXiv.2305.20009.
[63] Jacob-Dolan C, Yu J, McMahan K, et al. Immunogenicity and protective efficacy of GBP510/AS03 vaccine against SARSCoV-2 delta challenge in rhesus macaques [J]. NPJ Vaccines, 2023, 8(1): 23.
[64] Shanehsazzadeh A, Bachas S, McPartlon M, et al. Unlocking de novo antibody design with generative artificial intelligence [EB/OL]. [2023-11-15]. https://doi. org/10.1101/2023.01.08.523187.
[65] Generate:Biomedicines. Our pipeline [EB/OL]. [2023-11-15]. https://generatebiomedicines.com/pipeline.
[66] Silverman AD, Karim AS, Jewett MC. Cell-free gene expression: an expanded repertoire of applications [J]. Nat Rev Genet, 2020, 21(3): 151-170.
[67] Dondapati SK, Stech M, Zemella A, et al. Cell-free protein synthesis: a promising option for future drug development [J]. BioDrugs, 2020, 34(3): 327-348.
[68] Lemieux J. Protein-protein interactions get a new groove on: adding a modern twist to a traditional assay [J]. Genet Eng Biotechn N, 2019, 39(11): 30-32.
[69] Porebski BT, Balmforth M, Browne G, et al. Rapid discovery of high-affinity antibodies via massively parallel sequencing, ribosome display and affinity screening [J/OL]. Nat Biomed Eng, 2023 Oct 09. [2023-11-15]. https://doi.org/10.1038/ s41551-023-01093-3.
[70] Tamasi MJ, Patel RA, Borca CH, et al. Machine learning on a robotic platform for the design of polymer–protein hybrids [J]. Adv Mater, 2022, 34(30): e2201809.
[71] Rapp JT, Bremer BJ, Romero PA. Self-driving laboratories to autonomously navigate the protein fitness landscape [EB/OL].[2023-11-15]. https://doi.org/10.1101/2023.05.20.541582.
[72] Yu T, Boob AG, Singh N, et al. In vitro continuous protein evolution empowered by machine learning and automation [J]. Cell Syst, 2023, 14(8): 633-644.
[73] AlQuraishi M, Sorger PK. Differentiable biology: using deep learning for biophysics-based and data-driven modeling of molecular mechanisms [J]. Nat Methods, 2021, 18(10): 1169-1180.
[74] Johnson SR, Fu X, Viknander S, et al. Computational scoring and experimental evaluation of enzymes generated by neural networks [EB/OL]. [2023-11-15]. https://doi. org/10.1101/2023.03.04.531015.
[75] Gasser HC, Oyarzun D, Rajan A, et al. Comparing a language model and a physics-based approach to modify MHC class-Ⅰimmune-visibility for the design of vaccines and therapeutics [EB/OL]. [2023-11-15]. https://doi. org/10.1101/2023.07.10.548300.
[76] Zhang Z, Xu M, Lozano A, et al. Physics-inspired protein encoder pre-training via siamese sequence-structure siffusion trajectory prediction [EB/OL]. [2023-11-15]. https://doi. org/10.48550/arXiv.2301.12068.
猜你喜欢
西安航空学院学报(2022年2期)2022-07-04
汽车零部件(2020年3期)2020-03-27
表面工程与再制造(2019年1期)2019-05-11
商界(2019年12期)2019-01-03
家庭影院技术(2018年9期)2018-11-02
IT经理世界(2018年20期)2018-10-24
通信电源技术(2018年3期)2018-06-26
军营文化天地(2018年1期)2018-02-10
小康(2017年16期)2017-06-07
学与玩(2017年12期)2017-02-16
