
重组荔枝类甜蛋白的高效表达、纯化及活性鉴定
2024-05-17 13:23曹琳彩文舜华王凯刘旭炜胡卓炎赵雷沈兴
食品与发酵工业 2024年9期
曹琳彩,文舜华,王凯,刘旭炜,胡卓炎,赵雷,沈兴
(华南农业大学 食品学院,广东 广州,510642)
荔枝(LitchichinensisSonn.)在世界各地的温暖气候地区广泛栽培[1],其果肉中含有多种营养成分,如多酚、多糖等,据报道对肝、心、脾等有诸多益处[2],然而部分人群一次大量食用荔枝果肉会导致“上火”等不良反应[3]。“上火”是源于中医学的一种概念,表现为局部发红、发热、疼痛,类似炎症或炎症性疾病[4]。这种食物不良反应对荔枝市场发展产生了影响。研究表明导致“上火”的食物含有促进炎症的成分,如HUANG等[5]将荔枝定义为“热性食物”并发现荔枝的水提取物增加了小鼠巨噬细胞RAW264.7中抗炎介质前列腺素E2(PGE2)的分泌与环氧合酶-2(cyclooxygenase 2,COX-2)的表达,WANG等[6]使用RAW264.7细胞对荔枝中的水溶性成分进行了筛选,明确了荔枝中水溶性蛋白可以增加RAW264.7细胞系中促炎介质如诱导型一氧化氮合酶(inducible nitric oxide synthase,iNOS)、COX-2、白介素-1β(interleukin-1β,IL-1β)以及抗炎介质血红素加氧酶(heme oxygenase,HO-1)的产生。由此可见,荔枝中水溶性蛋白质是导致荔枝炎症性“上火”的主要原因。本团队前期研究发现,利用荔枝中分离纯化得到的类甜蛋白(litchi thaumatin-like protein,LcTLP)刺激RAW264.7细胞,会显著提高iNOS和COX-2的基因表达量以及炎症因子分泌量[7],继而推测LcTLP是食用荔枝引起炎症反应的主要原因,但目前LcTLP分子晶体结构的解析以及其在细胞生命活动中的功能机制等还没有深入的研究,因此大量的高纯度的活性LcTLP获取是急待解决的前提。
LcTLP可从新鲜果实中提取制备,但其产量低、周期长且纯度不高[7],而重组表达则提供了更优的解决方案。有研究对TLP家族中的其他来源进行了异源表达实验,但仍然没有一个高效的方案,如MERVAT 等[8]通过使用载体pET-28a(+)在大肠杆菌中表达番茄TLP(NP24),证明该基因在大肠杆菌中作为包涵体过度表达。此外,余宇等[9]对小麦TLP构建的TLP-pET-32a载体在表达菌株BL21(DE3)中也为包涵体表达。因此,为避免LcTLP的原核表达形成包涵体,本研究采用融合标签技术为大量表达可溶性LcTLP提供了解决途径。NusA标签(N-utilization substance A tag)能够提高E.coli表达重组蛋白的溶解度和稳定性,如ZACHARCHENKO等[10]使用NusA标签对人类蛋白磷酸酶1进行了高效的可溶表达。此外,适宜的纯化策略也是获得高纯度、有活性的重组蛋白的必要前提。因此本研究采用融合NusA标签和多组氨酸标签(His6-tag)构建了重组LcTLP表达载体,对表达菌株进行了优化。此外,还建立了两步法纯化方案,对纯化后的蛋白进行细胞炎症活性实验,为后续研究LcTLP在细胞生命活动中的功能机制奠定了实验基础。
1 材料与方法
1.1 材料与仪器
原核表达载体pCold-NusA-thrombin site、克隆菌株E.coliDH5α,广州艾迪生物科技有限公司;小鼠单核巨噬细胞白血病细胞RAW264.7,中国科学院细胞库;TIAN prep Mini Plasmid Kit、表达菌株BL21(DE3)pLysS、BL21(DE3)C43、BL21(DE3)感受态细胞,天根生化科技有限公司;BamH I、Hind Ⅲ内切酶、Fast Digest限制性内切酶,赛默飞世尔科技(中国)有限公司;AxyPrep DNA凝胶回收试剂盒,北京雅安达生物技术有限公司;AGE预混胶试剂盒,英国gene fist公司;乙二胺四乙酸(ethylene diamine tetraacetic acid,EDTA)、咪唑、溶菌酶、蛋白酶抑制剂100X、NO试剂盒,上海碧云天生物技术有限公司;低分子质量蛋白质marker、DNA连接试剂盒,德国Takara公司;全能核酸酶,翌圣生物科技(上海)股份有限公司;胰蛋白胨、酵母提取物、琼脂粉,英国Oxoid公司;100%Triton,Sigma公司;凝血酶,北京索莱宝科技有限公司;MTT细胞毒性试剂盒,南京建成生物工程研究所;其他化学试剂均为国产分析纯。
PCR仪、Nano Drop 2000/2000C分光光度计,美国赛默飞公司;恒温培养箱,日本三洋公司;SW-CJ-2FD洁净工作台,苏州安泰空气技术有限公司;SHA-CA数显水浴恒温振荡器,常州荣华仪器制造有限公司;光栅型酶标仪,美谷分子仪器有限公司;高速大容量离心机,美国贝克曼库尔特公司;蛋白质层析系统(AKTA start)、His trap FF、Hiload 16/600 superdex 75pg、超滤浓缩离心管,美国思拓凡公司;垂直电泳装置、全能型成像系统,美国伯乐公司。
1.2 实验方法
1.2.1 原核表达载体的构建
使用云舟生物在线网站对LcTLP基因(GenBank登录号JF682821)进行大肠杆菌密码子优化后,合成并插入pCold-NusA-thrombin site表达载体。将连接产物转化至E.coliDH5α感受态细胞中,将菌液涂于氨苄平板上,在37 ℃下培养12 h,挑取单克隆菌落接入含100 μg/mL氨苄青霉素LB培养基中,以220 r/min的转速振荡,37 ℃培养14 h后提取质粒,经测序鉴定正确的重组质粒命名为pCold-NusA-LcTLP。
1.2.2 重组蛋白诱导表达
分别取1 μL pCold-NusA-LcTLP质粒加入至BL21(DE3)pLysS、BL21(DE3)C43、BL21(DE3)感受态细胞中。采用热激法进行重组质粒的转化,冰上孵育30 min,42 ℃热激45 s,在冰上孵育3 min。接着加入950 μL预热的LB培养基,37 ℃摇床孵育60 min。孵育结束后离心去除上清液重悬涂板。将菌液涂布在相应抗性的LB平板上,37 ℃恒温培养16 h。挑取单菌落至LB抗性培养基,37 ℃、200 r/min培养过夜。加入1%(体积分数)扩增菌液于TB抗性培养基中,37 ℃、200 r/min条件下培养3 h后加入异丙基-β-D-硫代半乳糖苷(isopropyl-β-D-thiogalactopyranoside,IPTG)至0.1 mmol/L,以未添加IPTG作为对照组,15 ℃、200 r/min继续培养20 h后终止。收集菌液进行离心,弃上清液,重悬于4×裂解缓冲液(0.3 mol/L NaCl、20 mmol/L Tris pH 8.0、10%(体积分数)甘油、0.5%(体积分数)Triton、1 mmol/L 3,5-二硝基水杨酸、1 mg/mL溶菌酶、1×蛋白酶抑制剂100X、2 mmol/L EDTA),于液氮和37 ℃水浴中反复冻融3次后,置于37 ℃、200 r/min恒温振荡30 min。然后加入5 mmol/L MgCl2溶液和25 U/mL全能核酸酶,4 ℃反应30 min后10 000 r/min离心30 min,取上清液过0.45 μm滤膜,进行后续纯化步骤。将收集的细菌裂解液上清液与诱导前对照组裂解液进行SDS-PAGE检测。
1.2.3 重组LcTLP的纯化
1.2.3.1 亲和层析纯化与酶切
采用5 mL His Trap FF镍亲和柱进行第一步纯化。首先用缓冲液A[0.5 mol/L NaCl溶液、20 mmol/L Tris pH 8.0、10%(体积分数)甘油、0.5%(体积分数)Triton]平衡3~5个柱体积后进行上样,设定3 mL/管进行收集。上样结束后,以10%(体积分数)缓冲液B(缓冲液A的基础上增加40 mmol/L咪唑)对柱子上结合的杂蛋白进行洗脱,然后提高咪唑浓度至200 mmol/L对目的蛋白进行洗脱。将相应的洗脱峰进行SDS-PAGE检测,收集目的蛋白溶液用NanoDrop测定其浓度,每1 mg蛋白样品中加入0.3 U凝血酶,37 ℃水浴反应2.5 h以切除NusA标签。
1.2.3.2 凝胶过滤层析纯化
采用Superdex75凝胶柱进行第二步纯化。首先用缓冲液(150 mmol/L NaCl溶液、20 mmol/L Tris pH 8.0)平衡1个柱体积,然后将上一步纯化得到的蛋白溶液用30 kDa V型超滤浓缩管浓缩至5 mL进行上样,设定3 mL/管进行收集,继续运行1.2个柱体积后结束程序,将收集到的洗脱峰进行SDS-PAGE检测,选取符合目的蛋白大小的洗脱峰留存备用。
1.2.4 Western blot检测
参照WANG等[11]报道的方法并略作修改。取纯化后重组LcTLP经SDS-PAGE后,以200 A恒压电流转移55 min至0.22 μm PVDF膜上,再将膜放入含50 g/L脱脂奶的TBST(Tris buffered saline with Tween-20)中,室温封闭1 h。加入1∶3 000(体积比)鼠抗His6标签单克隆抗体,4 ℃过夜后用TBST洗膜3次,再加入1∶5 000(体积比)的HRP标记的山羊抗鼠IgG二抗,室温反应1 h,用TBST洗膜3次,最后加入超敏发光液,在凝胶成像系统成像,检测目的蛋白的表达情况。
1.2.5 重组LcTLP产量与纯度测定
重组LcTLP的纯度使用Image Lab软件进行分析[12]。取每1 L培养基中提取过程中裂解上清液中的总蛋白、NusA-thrombin-LcTLP、重组LcTLP的浓度根据BCA试剂盒的方法检测并根据浓度与该提取步骤的体积进行换算得到蛋白质总量。将BCA试剂A与试剂B按50∶1(体积比)比例混合均匀,制成BCA工作液。25 mg/mL蛋白标准溶液用10 mmol/L K3PO4缓冲液分别稀释质量浓度为0.05、0.1、0.2、0.3、0.4、0.5 mg/mL的标准品,同时样品组稀释多种不同的浓度。在标准品和样品中加入200 μL的BCA工作液,37 ℃孵育30 min,使用酶标仪测定562 nm处的吸光度,制作标准曲线y=0.631 2x+0.185 9计算样品蛋白的浓度。
1.2.6 重组LcTLP的生物活性鉴定
1.2.6.1 细胞培养与模型建立
将小鼠RAW264.7细胞在含体积分数10%胎牛血清、1%非必需氨基酸、1%谷氨酰胺和1%青霉素-链霉素双抗液(均为体积分数)的DMEM培养基中培养(37 ℃、5% CO2),取对数生长期细胞用于进一步实验。空白对照组加入等体积的培养基,阳性对照组加入1 μg/mL的LPS,样品组分别加入用培养基稀释为1、2 μg/mL质量浓度的重组LcTLP。
1.2.6.2 细胞存活率的测定
细胞存活率测定参照刘素贞等[13]的方法。取对数生长期细胞,以1×105个/孔的密度将细胞接种在96孔细胞培养板中培养12 h使细胞完全贴壁,移去培养基后分别加入1 μg/mL质量浓度的LPS、1 μg/mL和2 μg/mL重组LcTLP进行对比。刺激细胞24 h后,移去细胞培养板上的溶液,在遮光条件下,向各孔中加入含CCK-8的培养液200 μL,在37 ℃培养箱中继续培养1 h后,使用酶标仪在450 nm处测定各孔吸光值。
1.2.6.3 细胞NO分泌量的测定
取对数生长期的RAW264.7细胞接种于96孔细胞培养板中,分别用1 μg/mL的LPS,1 μg/mL和2 μg/mL重组LcTLP培养24 h后,取上清液使用试剂盒测定NO的分泌量。
1.3 数据处理
在SPSS软件上使用单因素方法分析对实验数据进行处理,P<0.05表示具有显著性差异。实验数据采用平均值±标准误差表示,使用Origin 2018软件进行图片绘制。
2 结果与分析
2.1 LcTLP表达载体构建与密码子优化
不同生物体的密码子使用方式存在的差异会影响到异源表达系统中目标基因的表达水平。密码子优化是通过克服与密码子使用和转移RNA(tRNA)丰度的物种特异性差异相关的限制来提高翻译率,增强蛋白表达与稳定性[14]。有效密码子数(effective number of codons,ENC)是评价基因整体密码子偏好性中最有参考价值的参数之一。ENC值描述的是某个基因的密码子偏好程度,ENC值越低,说明密码子使用偏好性越强[15]。以GenBank公布的LcTLP基因为模版,进行密码子优化,前后对比如图1-a所示。通过EMBOSS在线网站对密码子优化前后ENC值和GC含量进行评估,优化前后的ENC值分别是52.56和47.15,GC含量无明显变化,分别为48.66%和50.67%,说明优化后的密码子更利于LcTLP在大肠杆菌中的可溶性表达。将优化后的LcTLP基因序列直接合成并插入载体pCold-NusA-thrombin site中,转化至E.coliDH5α感受态中得到转化子,通过扩大培养提取重组质粒,质粒图谱见图1-b。该质粒中,NusA标签基因与LcTLP基因之间含凝血酶酶切位点,LcTLP基因另一端则含有His6标签基因序列,经测序后表明该载体构建成功。

a-序列对比图;b-质粒图谱
2.2 重组蛋白NusA-LcTLP的表达优化
本研究中初步采用了3种不同的菌株对重组蛋白NusA-LcTLP进行诱导表达,结果如图2-a所示,可以看出3种菌株表达的重组蛋白含量均不高,但BL21(DE3)pLysS菌株表达裂解后的背景更为干净,杂蛋白比例低。BL21(DE3)pLysS、BL21(DE3)C43和BL21(DE3) 3种不同的大肠杆菌宿主细菌在使用T7启动子时具有不同的蛋白质表达水平[16-18]。其中,BL21(DE3)pLysS菌株含有T7溶菌酶的基因,该基因作用于大肠杆菌细胞壁中的肽聚糖以溶解细菌,减少目标基因的背景表达却不干扰IPTG诱导的表达,使其更适合表达有毒蛋白[19]。相比其他2种基因型的菌株,该基因型菌株对降低目的基因的表达背景水平效果最好,有利于简化后续纯化蛋白的步骤,提高蛋白纯度,因此选用BL21(DE3)pLysS菌株作为表达菌株进行表达条件的优化。图2-b为优化后的表达效果,可以看出裂解菌体离心后的上清液在80 kDa左右有明显的NusA-LcTLP特征蛋白条带出现,其中NusA分子质量为54.87 kDa。His6标签分子质量为0.81 kDa,LcTLP含223个氨基酸残基,分子质量约为24 kDa,因此NusA-LcTLP理论分子质量约为80 kDa。作为对照的未诱导重组菌体中无该蛋白条带,说明成功表达得到了NusA-LcTLP重组蛋白。
2.3 NusA-LcTLP亲和层析纯化结果
由于目的蛋白NusA-LcTLP带有His6标签,首先采用Ni-NTA进行亲和层析纯化。收集不同咪唑浓度缓冲液洗脱得到的组分,进行SDS-PAGE检测(图3)。结果显示,在使用不含咪唑的缓冲液A以及含40 mmol/L咪唑的洗脱液冲柱时,洗脱液中仅含少量目的蛋白,而使用含200 mmol/L的咪唑洗脱时可得到较纯的NusA-TLP,表明通过此方法可以较有效地初步分离NusA-LcTLP。收集200 mmol/L咪唑洗脱得到的目的蛋白进行后续酶切实验将NusA标签与重组LcTLP分离。
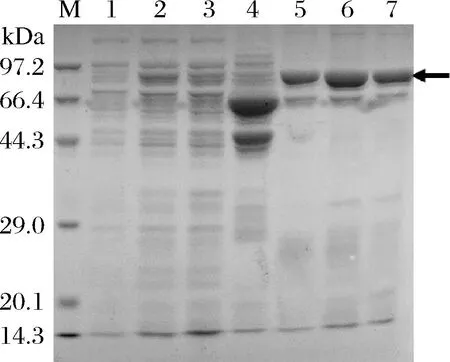
图3 NusA-LcTLP亲和纯化产物SDS-PAGE图Fig.3 NusA-TLP affinity purification product SDS-PAGE analysis
2.4 NusA-LcTLP酶切与Western blot验证
采用凝血酶对NusA标签与重组蛋白进行切割分离,从图4-a可以看出,酶切后NusA-LcTLP中的NusA标签和重组LcTLP被成功切割分开,电泳中出现2条相应大小的条带。重组LcTLP在SDS-PAGE结果中出现在29 kDa附近,略高于计算分子质量24 kDa。原因可能是由于His6标签中包含多个带正电荷的组氨酸,因此目标蛋白的电荷状态发生了改变,进而影响了它在SDS-PAGE中的表现,使其分子质量的测量结果偏大[20]。图4-b Western blot结果显示,酶切前免疫印迹最深处于80 kDa处,酶切后该位置的免疫印迹消失,24 kDa处出现了较深的免疫印迹,可以说明酶切成功。
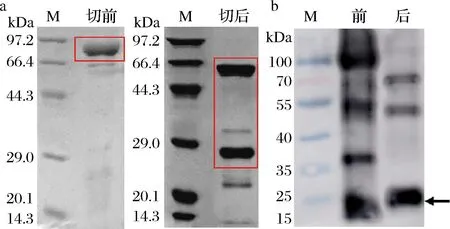
a-SDS-PAGE图;b-Western blot图
2.5 凝胶过滤层析分离重组LcTLP
通过图5可以看到,酶切产物经过凝胶过滤层析纯化后得到2个洗脱峰,将不同的洗脱峰收集起来进行SDS-PAGE检测。2个洗脱峰分别对应为泳道1为NusA标签,泳道2为重组LcTLP。说明NusA标签与重组LcTLP两个蛋白可以很好地分离。电泳结果显示,纯化后的重组LcTLP条带单一,经Image Lab软件分析显示纯度达90%。

a-凝胶过滤层析分离酶切产物峰图;b-SDS-PAGE图
2.6 BCA法测定蛋白浓度与产量
如表1所示,经IPTG诱导后,每1L TB培养基中湿菌体裂解后离心上清液所得总蛋白含量为2 711.03 mg,经第1次纯化后可得NusA-LcTLP 286.95 mg。通过酶切与凝胶过滤层析纯化之后重组LcTLP总得量为36.26 mg。在CHEN等[7]的研究中,从100 g新鲜荔枝果肉中分离出果肉蛋白后经丙酮沉淀和PBS提取,使用不同饱和度的(NH4)2SO4沉淀进一步透析与冻干收集,可得纯度为70%的4.5 mg LcTLP。与此相比,重组LcTLP的纯度远高于化学法提取的LcTLP。而FUCHS等[21]经过多重化学纯化在120.2 kg成熟樱桃中获得约1 mg高度纯化的樱桃TLP(nPru av 5),但发现此法提取的nPru av 5上存在碳水化合物部分,因此该提取方法也无法运用于蛋白晶体结构解析的实验中。FUCHS等[21]还对nPru av 5进行原核表达发现为包涵体表达,经变性纯化后得重组nPru av 5产量为0.7 mg/L培养基。除nPru av 5外,多种TLP在原核表达中形成包涵体被报道,如番茄TLP[8]、小麦TLP[9]和百合TLP[22]等。综上所述,本实验通过助溶标签载体的构建、密码子和表达菌株的优化得到了重组LcTLP 36.26 mg/L培养基,实现了重组LcTLP在原核系统的高效可溶性表达,并成功建立了一种高纯度的制备方法。

表1 BCA法测定蛋白浓度与产量Table 1 Determination of protein concentration and total amount by BCA method
2.7 重组LcTLP的生物活性鉴定
图6显示了不同浓度的LPS、重组LcTLP对RAW264.7细胞存活率和NO分泌水平的影响。由图6-a可知3组样品的细胞存活率均在100%以上,说明1 μg/mL LPS、1 μg/mL和2 μg/mL重组LcTLP对RAW264.7细胞均无毒害作用且有一定的促进细胞生长的效果,该剂量可以用于后续实验。
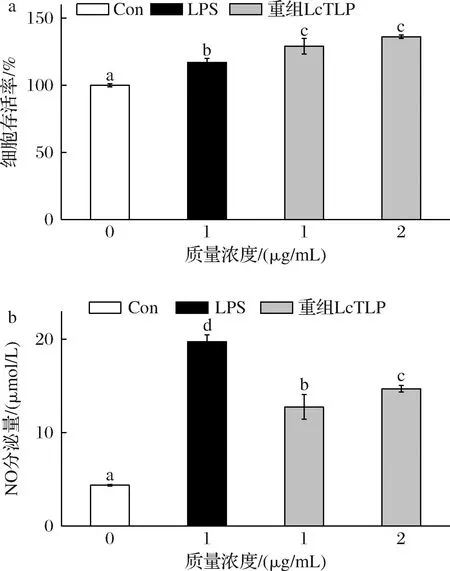
a-细胞存活率;b-NO分泌量
RAW264.7细胞来自小鼠巨噬细胞系,它们在控制免疫反应和炎症方面发挥着重要作用。NO是炎症过程中的重要信号分子[23],在机体内通常存在正常水平的NO,当巨噬细胞被激活时,它们会释放高水平的NO并激活 NF-κB信号通路,从而导致促炎因子的分泌并引发炎症反应,因此NO含量可用作炎症水平的衡量[24]。由图6-b可知,NO的分泌量随着重组LcTLP的浓度升高均呈现剂量依赖性增加,阳性对照LPS组含量为19.74 μmol/L,与空白组有显著差异,可以说明细胞炎症造模成功。在1~2 μg/mL质量浓度范围内,重组LcTLP的NO分泌量分别为12.75和14.68 μmol/L,是空白组的2.91倍和3.36倍,以上组分都显著提高了细胞NO的分泌。在WANG等[25]的研究中,10 ng/mL LPS和荔枝果实中天然提取的LcTLP刺激RAW264.7细胞NO分泌量约分别为50和30 μmol/L,约为空白组的10和6倍。可以说明与天然提取LcTLP一样,重组LcTLP具有增强RAW264.7细胞分泌NO的效果,具有一定的促炎活性,为后续提供了实验基础。
3 结论
本研究通过密码子优化、构建含助溶标签NusA标签和用于亲和层析的His6标签原核表达载体、表达菌株优化,建立了一种高效表达可溶性重组LcTLP的方法。经原核表达的结果分析最合适的表达菌株为BL21(DE3)pLysS。使用亲和层析技术、酶切技术和凝胶色谱层析技术对原核表达的重组蛋白进行纯化,经蛋白电泳与免疫印迹鉴定,证明成功高效表达并纯化得到可溶性重组LcTLP,其产量为36.26 mg/L,纯度达90%以上。此外,RAW264.7细胞炎症活性验证表明,经原核系统表达纯化的LcTLP对RAW264.7细胞无毒害作用且能够促进RAW264.7中NO的分泌量并呈现一定的浓度依赖性,说明了重组LcTLP导致巨噬细胞产生炎症反应。证实了重组LcTLP具有一定的生物活性,可为后续蛋白结构解析与功能研究奠定基础。
猜你喜欢
食用菌(2023年6期)2023-11-28
草地学报(2022年3期)2022-03-28
今日农业(2021年11期)2021-11-27
生物学通报(2020年11期)2020-10-22
车迷(2018年11期)2018-08-30
中成药(2018年7期)2018-08-04
海峡姐妹(2018年3期)2018-05-09
公民与法治(2016年10期)2016-05-17
计算机工程(2015年8期)2015-07-03
生命科学研究(2014年1期)2014-04-29
