
散发性包涵体肌炎1例☆
2024-05-06 07:21:48林晓丹陈龙郑富泽郑颖曾明慧林锋林珉婷王柠王志强
中国神经精神疾病杂志 2024年2期
林晓丹 陈龙 郑富泽 郑颖 曾明慧 林锋※ 林珉婷※ 王柠※ 王志强※
散发性包涵体肌炎(sporadic inclusion body myositis,sIBM)是一种缓慢进展的炎症性肌病,多见于50 岁以上,主要表现为股四头肌和指屈肌的无力、萎缩[1]。病理特征主要包括T细胞为主的炎症浸润和退行性改变。sIBM 的确诊依赖于独特的临床表型和组织病理学,但疾病早期表现可不典型,易导致误诊和漏诊,使sIBM 的早期诊断成为本病争议话题[2]。目前国内sIBM 的报道相对较少,尤其是关于TDP-43(TAR DNA binding protein-43)、p62(Sequestosome-1)蛋白聚集及cN1A(cytoplasmic 5'-nucleotidase 1A)抗体方面的应用研究更少。福建医科大学附属第一医院确诊sIBM患者1例,现报告如下。
1 临床资料
患者,男,69 岁,因“渐进性四肢无力、萎缩3 年余”于2022年12月就诊我院。患者2019年发现双下肢无力,表现登梯、爬坡、蹲立困难,伴大小腿肌肉萎缩,逐渐累及双上肢,出现握物、持物差,呈不对称受累,无吞咽困难、言语含糊,无肌痛、肌跳等。2021 年4 月吞咽费力,无饮水呛咳,门诊查肌电图提示肌源性损害。既往体健,否认相关肌肉疾病家族史。
体格检查:神志清楚,眼球活动正常,声音稍含糊,吞咽稍费力,咽反射存在,余脑神经查体未见异常。颈屈、伸肌力5 级,左侧腕伸肌力4+级,右侧腕伸肌力4 级,左侧腕屈肌力4级,右侧腕屈肌力4-级,双侧指伸肌力4-级,双侧指屈肌力3+级,左侧髂腰肌力5-级,双侧股四头肌力5-级,左侧腘绳肌力4+级,右侧腘绳肌力5-级,双侧胫前肌力4级,余肢体肌力正常。双下肢远端肌萎缩,四肢肌张力正常,步态正常。深浅感觉未见异常。四肢腱反射对称迟钝,病理征阴性,共济运动正常。洼田饮水试验阴性。
辅助检查:血清肌酸激酶(CK)774 U/L↑(参考值:50~310 U/L),乳酸脱氢酶325 U/L↑(参考值:120~250 U/L),肌酸激酶同工酶44 U/L↑(参考值:<25 U/L),血清肌红蛋白、甲状腺功能、肿瘤标记物、抗核抗体谱、神经元抗原IgG抗体谱未见异常。神经电生理:双侧尺神经部分损害,双下肢运动复合肌肉动作电位(compound muscle action potiential,CMAP)波幅低;针电极:肌源性损害。双大腿肌肉MRI:双侧股外侧肌、股内侧肌、股中间肌、大收肌、缝匠肌、股薄肌、竖脊肌存在脂肪浸润,远端受累重于近端;股直肌、半膜肌、半腱肌无明显脂肪浸润,臀肌无明显受累;双侧股直肌、大收肌、股薄肌部分炎症浸润(图1)。膈肌彩超:双侧膈肌运动减低程度,左侧膈肌运动相对位移。心脏彩超、肺功能未见明显异常。
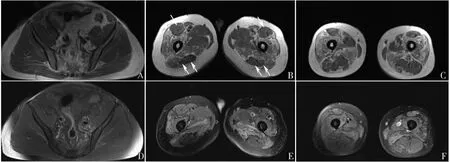
Fig.1 Patient's bilateral thigh muscle MRI图1 患者双侧大腿肌肉MRI A、D,臀部;B、E,大腿上段;C、F,大腿下段,箭头为股直肌、半膜肌、半腱肌(T1WI序列:A、B、C,T2WI-FS序列:D、E、F)。
常规组化病理:HE 染色可见肌纤维中度大小不一,部分肌纤维内可见空泡样结构,内核纤维轻度增多,少量坏死肌纤维,偶见再生肌纤维,间质纤维结缔组织轻度增生,部分肌纤维间隙伴炎症细胞浸润,并偶见浸润非坏死肌纤维,未见明显脂肪细胞;改良Gomori(MGT)染色可见部分肌纤维内不规则镶边空泡(rimmed vacuoles,RVs),未见破碎红肌纤维及异常物质沉积;还原型辅酶Ⅰ(NADH-TR) 染色可见部分酶活性缺失肌纤维,未见分叶样、虫噬样及靶样肌纤维;细胞色素C 氧化酶(COX)/琥珀酸脱氢酶(SDH)染色偶见蓝纤维;三磷酸腺苷酶 (ATP) 染色可见两型肌纤维呈镶嵌排列,Ⅰ型约占 80%(pH=4.3);酸性磷酸酯酶(ACP)染色阳性;过碘酸盐(PAS)、油红O(ORO)染色未见糖原、脂滴增多(图2A~G)。免疫组化病理:主要组织相容性复合物-I(majorhistocompatibilitycomplex-Ⅰ, MHC-Ⅰ)弥漫表达于肌纤维表面,部分肌纤维间隙可见CD3、CD8、CD68呈片状浸润,并围绕非坏死肌纤维浸润,p62蛋白在含有RVs的肌纤维内散在聚集,CD20 染色阴性(图2H~L)。电镜下可见肌纤维肌丝间中等量涡轮状髓样小体,肌丝灶性溶解,未见明显管丝状包涵体(图3A)。免疫荧光染色:TDP-43蛋白在肌细胞核、胞质内散在聚集(图4)。肌炎抗体谱23 项半定量示:抗cN1A 抗体阳性(图3B),余抗体阴性。患者最终诊断为sIBM,治疗上予以维生素B6、维生素B1、辅酶Q口服。1年后随访,患者四肢无力、吞咽费力,较前稍进展。

Fig.2 Histopathology of the patient's skeletal muscle biopsy图2 患者骨骼肌组织病理染色 A,肌纤维大小不一,可见空泡样结构,内核纤维增多,伴少量炎症细胞浸润,并偶见浸润非坏死肌纤维(HE×100);B,蓝纤维(箭头, COX/SDH×100);C, 部分酶活性缺失肌纤维(箭头, NADH×100);D,两型肌纤维呈镶嵌排列(ATP×100);E,空泡样结构(箭头,HE×400);F: 镶边空泡(箭头, MGT×400);G, ACP 阳性(箭头,×400);H: 肌膜弥漫表达(箭头,MHC-Ⅰ×400);I~K: CD3、CD8、CD68 阳性(箭头,×400);L:肌浆内聚集(箭头,p62×400)。

Fig. 3 Patient's muscle electron microscopy and cN1A antibody testing图3 患者肌肉电镜图与cN1A抗体检测 A:涡轮状髓样小体(箭头,×2000);B:cN1A抗体(+)
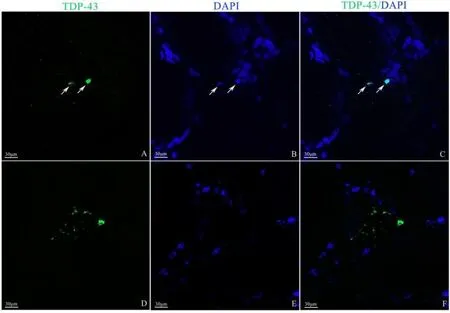
Fig.4 Immunofluorescence staining of TDP-43 in the skeletal muscle of patient图4 患者骨骼肌TDP-43蛋白免疫荧光染色 A~C:胞核聚集(箭头,×400);D~F:胞质内聚集(×400)。
2 讨论
sIBM 经过多次修订,在第188 届欧洲神经肌肉中心(European Neuromuscular Centre, ENMC)国际研讨会上制定了新的分类标准[3],将其分为临床病理确诊、临床确诊和可能诊断,本例患者属于临床病理确诊的sIBM。
sIBM早期没有典型特征,常被误诊为坏死性肌炎、多发性肌炎、神经元核内包涵体病(neuronal intranuclear inclusion disease,NIID) 或其他伴有RVs 的遗传性肌炎(hereditary IBM,hIBM)。坏死性肌炎、多发性肌炎病理学上均提示肌纤维坏死和再生,但一般不伴有包涵体。sIBM 和NIID 肌纤维内都可见包涵体,但NIID主要累及运动神经元,肌电图提示神经源性损害,颅脑MRI 提示皮髓交界处弥散加权成像(diffusion-weighted imaging,DWI)高信号。sIBM 和hIBM常被认为具有相似临床和病理特征的同一种疾病呈散发性和遗传性的不同遗传方式,实际上是具有不同临床表型和患病率的两种疾病,后者的病理表现缺少独特的炎症细胞浸润。因此对于主诉膝、足无力、大腿萎缩、无感觉异常的中老年患者,要注意与sIBM 鉴别。本例患者隐匿起病,进展缓慢,受累肌群呈不对称分布,股四头肌、腕屈肌、指屈肌无力较突出,符合sIBM经典起病模式。部分sIBM患者可出现吞咽困难,长期吞咽困难可出现营养缺乏、体质量减轻和吸入性肺炎等并发症,是导致sIBM 死亡的主要原因[4]。本例患者亦出现延髓肌轻度受累,建议早期进行吞咽肌功能训练。
slBM 患者CK 小于正常上限的15 倍[3],本例患者CK 轻度升高。EMG 在sIBM 的确诊中没有明确意义,但早期可判断是否存在肌源性损害,排除运动神经元病变或多发性周围神经病变。部分sIBM 患者可出现强直电位,特别是在病变严重的肌群中,研究发现强直电位与炎症浸润的严重程度相关[5],本例患者合并神经源性损害,未检测到强直电位。sIBM 肌肉MRI 遵循与临床表型相同的模式,称为“影像学标准”,优先累及股四头肌和指屈肌,包括脂肪浸润和短T1反转恢复序列(short T1inversion recovery,STIR) 高信号,早期股直肌通常不受累,多呈不对称分布,存在由近端向远端进展的趋势,远端肌群受累最明显。小腿部受累最多的是内侧腓肠肌,而胫骨后肌和比目鱼肌相对保留[6]。与其他炎性肌病相反,sIBM患者肌群的脂肪浸润通常比水肿更突出。本例患者双侧大腿MRI符合sIBM的影像学标准(图1)。
病理学特征是sIBM 诊断的金标准,包括:肌膜内炎症浸润、RVs、蛋白聚集、15~18 nm 管丝包涵体[3]。其中肌膜内炎症是以CD8+T 淋巴细胞为主的炎性细胞浸润非坏死肌纤维;蛋白聚集包括淀粉样蛋白、p62、SMI-31 (sternberger monoclonals incorporated-31)及TDP-43 等蛋白沉积物,p62、TDP-43 蛋白沉积物在非sIBM 自身免疫性肌病中罕见[7]。此外,经常观察到肌纤维表面MHC-I 过表达,COX 阴性纤维,异常的线粒体变化,如破碎红纤维等。虽然单独的病理改变是非特异性的,但出现在同一个体中,支持诊断sIBM。本例患者符合sIBM 的组织病理学诊断,肌肉组织在电镜下未见管丝包涵体,可能与取材受限未切到存在包涵体的层面,也可能与病程较短有关。
sIBM 的发病机制尚不清楚,越来越多的证据提示sIBM是一种自身免疫性T 细胞介导的疾病[8]。部分sIBM 患者血清中可检测到自身抗体,以抗核抗体、类风湿因子、抗心磷脂抗体、抗Ro 抗体最常见。本例患者抗核抗体谱、神经元抗原IgG抗体谱均未见异常,但cN1A抗体阳性。cN1A抗体是存在于部分sIBM 患者血清中的一种自身抗体,对于诊断sIBM 灵敏度有限,约为30%~50%,具有高特异性,通常超过90%[9-10]。目前,sIBM 的诊断主要依靠病理活检,但一些病理特征仅在疾病后期,严格根据这些病理特征将漏诊早期患者,可能错过最佳的干预时段,因此寻找新型生物标志物尤为重要。
目前国内sIBM 的报道相对较少,尤其是鲜有关于TDP-43、p62 蛋白聚集及cN1A 抗体等潜在生物标志物的应用,期望通过本病例的探讨及文献复习能加深临床对生物标志物的认识。临床实践中,常规的激素和免疫抑制剂等治疗可能会减轻炎症,在治疗的初始阶段有所成效,但是否能阻止疾病的进展,仍未可知,需要更多大型、长期的临床试验证明。
猜你喜欢
中国临床医学影像杂志(2022年6期)2022-07-26 07:17:24
中国临床医学影像杂志(2022年5期)2022-07-26 07:11:54
心电与循环(2021年4期)2021-11-29 02:41:56
国际放射医学核医学杂志(2021年10期)2021-02-28 08:43:54
心电与循环(2020年3期)2020-06-18 13:43:12
天津农学院学报(2016年2期)2016-12-01 05:40:05
中国眼镜科技杂志(2016年17期)2016-10-24 08:36:30
作文周刊·小学一年级版(2015年46期)2015-07-06 10:51:17
数学大世界·小学低年级辅导版(2009年8期)2009-07-28 08:00:12
学生天地·小学低年级版(2009年5期)2009-06-29 09:57:32
