
铍团簇吸附一氧化碳分子的密度泛函理论研究
2024-04-28 05:37林莉邝向军
西南科技大学学报 2024年1期
林莉 邝向军


摘要:采用密度泛函理論和广义梯度近似方法,在 Perdew - Wang 91水平上对 Be n CO( n =2~13)团簇进行了全电子计算。结果表明:CO 分子倾向于平行吸附在纯 Be n 团簇的表面并占据铍团簇的外部位置,吸附后主团簇结构基本没有发生较大的突变,但是 CO 分子的吸附会使 Be n 团簇的对称性下降,n =6 , 8时铍团簇对 CO 分子的吸附相对较强,n =4 , 7 , 11时铍团簇对 CO 分子的吸附相对较弱;吸附 CO 分子后,多数 Be Be键长以及 C O 的键长变长,即一氧化碳的吸附会使 Be Be以及 C O 的相互作用减弱;吸附之后 Be n CO 团簇的能隙、垂直电离势、垂直亲和势均发生了较为明显的变化,Be n CO 的能隙出现了较为明显的奇偶振荡并在 Be8 CO 出现了最大值,吸附前后团簇的垂直电离势、垂直亲和势曲线变化大趋势基本相同,随着原子数的增加团簇的垂直电离势逐渐降低而垂直亲和势逐渐升高。CO 分子吸附后对纯 Be n 团簇的稳定性产生了较大影响,C O 的键长、Be Be键长及团簇的能隙均发生了较大变化,吸附后仍存在幻数结构 Be8 CO 。
关键词:铍团簇一氧化碳分子吸附密度泛函理论
中图分类号:O641 文献标志码:A 文章编号:1671-8755(2024)01-0044-07
Density Functional Theory Study on the Adsorption of Carbon Monoxide Molecule by Beryllium Clusters
LIN Li , KUANG Xiangjun
(School ofMathematics and Physics , Southwest University ofScienceand Technology , Mianyang 621010 , Sichuan , China )
Abstract: An all-electron calculation of Be n CO ( n =2 -13) clusters have been performed by using density functional theory with the generalized gradient approximation at the Perdew - Wang 91 level . The results reveal that CO molecule tends to be adsorbed horizontally on the surface of pure Be n clusters and occupies the outer positions of beryllium clusters . After adsorption , the structure of the main clusters does not change greatly , but the adsorption of CO molecule will reduce the symmetry of Be n clusters . When n is 6 , 8 , the adsorption of beryllium clusters to CO molecule is relatively strong , and when n is 4 , 7 , 11 , the adsorption of beryllium clusters to CO molecule is relatively weak . After adsorption of CO molecule , most of the Be Be bonds and the C O bond become longer , that is , the adsorption of carbon monoxide weakens the interactions of Be Be and C O . After adsorption , the HOMO - LUMO Gap , vertical ioniza- tion potential and vertical electron affinities of Be n CO clusters change obviously. The HLG of Be n CO shows obvious odd - even oscillation and the maximum value appears at Be8 CO . The change trends of VIP and VEA curves of clusters before and after adsorption are basically the same . With the increase of thenumber of atoms , the VIP of clusters decreases while the VEA of clusters increases gradually. The adsorp- tion of CO molecule has a great influence on the stability of pure Be nclusters . The C O bond length , the Be Be bond length and HLG of clusters change greatly. The magic number structure Be8 CO still exists after adsorption .
Keywords : Beryllium clusters; Carbon monoxide molecule; Adsorptions; Density functional theory
近年来,随着团簇学科的迅猛发展,研究者对团簇的各种物理和化学性质以及它们随着团簇分子大小的演變规律已经有了较为系统的认知。人们希望通过在团簇表面吸附不同的原子、分子来合成具有独特性质的新型团簇材料,这将对催化科学、表面科学、材料科学、凝聚态物理等领域产生深刻的影响。金属团簇作为团簇的重要组成部分,其在理论及实验上是人们关注的热点。二价碱土金属铍是一种稀有的金属材料,也是最轻的金属之一。在宏观上,金属铍是航天、航空、原子能及冶金工业必不可少的材料[1]。从微观上看,铍也有着一些有趣的特性,二聚体铍是弱结合,而较大的铍团簇表现为自由电子体系并显示金属性质[2-5]。Wang 等指出,小分子铍原子之间的相互作用类似于范德瓦耳斯相互作用,随着团簇尺寸的增加,铍原子之间的成键会转换成共价键,再进一步转变成金属键[6]。此外,Kawai 等发现当 n =4 , 10 , 17时铍团簇中出现了幻数簇[7] , 这与电子壳模型的推论是一致的[7-9]。铍原子除了本身的特殊性之外,其另一重要用途是以铍原子为主团簇掺杂其他金属,进而形成二元合金。葛桂贤[10-12]对铍团簇掺杂 Na , Mg , Fe , C o , Ni 等金属进行了研究,结果表明掺杂后团簇的结构及稳定性都会发生显著的变化;雷雪玲等[13]分析了 Ben +1与 Be n Li 的基态结构、稳定性以及极化率,结果表明锂原子的掺杂增加了团簇的极化率。这些研究表明,金属原子的掺杂对铍团簇结构、能量、稳定性都产生了较大的影响。在过去的研究中,人们更多讨论了金属掺杂对铍团簇的影响,而对于铍团簇吸附非金属的研究较少。事实上,铍原子可以和许多非金属相互作用形成多元化合物,例如氧化铍(BeO) , 尽管它具有较大的毒性,但它可以用于制造铍氧化物、陶瓷、玻璃,也可用作核反应器燃料和减速剂,还可用作有机反应催化剂[14]。李淑萍等[15]发现铍团簇表面存在大量悬挂键,能够更好吸附一氧化氮分子,进而减轻一氧化氮排放对空气的污染。董兰等[16]对铍团簇吸附氢气进行了研究,发现随着铍团簇原子数量增加,其与氢原子的杂化程度逐渐变大。尽管越来越多的研究者投入于对该领域的研究,但有关 CO 在铍团簇表面的吸附行为的研究未见报道。小分子铍团簇不仅具有复杂的表面,且原子之间的相互作用较弱,这与其他金属有着明显的区别,因此研究铍团簇吸附一氧化碳的结构、能量、吸附位置、吸附强度具有重要意义。本文从第一性原理出发,在 Perdew - Wang 91( PW91)水平上运用广义梯度近似方法对一氧化碳分子在铍团簇上的吸附进行计算,并对以下问题进行分析:(1)一氧化碳的吸附是否会对纯 Be n 团簇的结构和稳定性产生影响;(2) Be n 团簇的结构和稳定性是如何演化的;(3)吸附后 CO 分子以及 Be n 团簇的键长、能隙如何变化;(4)吸附后团簇是否仍存在幻数结构。
1 计算方法
在计算过程中,采用 Materials Studio 中的 DMol3模块,选择广义度近似( GGA)方法进行拟合,选用电子的交换关联函数( PW91)进行全电子计算,SCF 容限为1.0×10-6 eV 。为了加速计算,使用迭代子空间中的直接反演( DIIS)方法,并且将拖尾值设置为0.005 Ha(1 Ha =2625.5 kJ·mol -1 , 下同)。在结构优化过程中,自旋不受约束,结构的对称性不受约束。最大力、能量和最大位移的收敛公差分别为0.05 eV/nm , 1.0 ×10-5 Ha 和0.05 nm 。在进行 Be n 团簇吸附一氧化碳分子的相关计算时,最关键的是确定最低能量结构。首先需要得到纯 Be n 团簇的最稳定结构。结合前人对纯 Be n 团簇的研究,发现密度泛函理论中的 PW91在纯 Be n 团簇的结构和稳定性上表现得非常好,因此笔者采用相同的方法构建纯 Be n 团簇并对其进行几何优化,优化结构如图1所示,与前人的研究成果一致[15]。在此基础上,为了得到 Be n CO 团簇的初始结构,让铍团簇的每个可能的非等价位置与 CO 分子成键,得到了 Be n CO 团簇的初始结构,包括3种可能的成键结构:C 原子与铍团簇成键、O 原子与铍团簇成键以及 C , O 原子同时与铍团簇成键。之后计算了吸附前后团簇的总能量( EBe )、平均结合能(ΔEBe )、振动频率、键长、最高已占据轨道能级、最低未占据轨道能级、能隙( HLG)、垂直亲和势( VEA)以及垂直电离势(VIP)等参数。
式中:ES 表示 EBe CO ( n =2~13)团簇的总能量;Eads表示 EBe nCO ( n =2~13)团簇的吸附能;E(Be nCO)+ 表示得到一个电子后团簇的总能量;E(Be nCO)-表示失去一个电子后团簇的总能量。
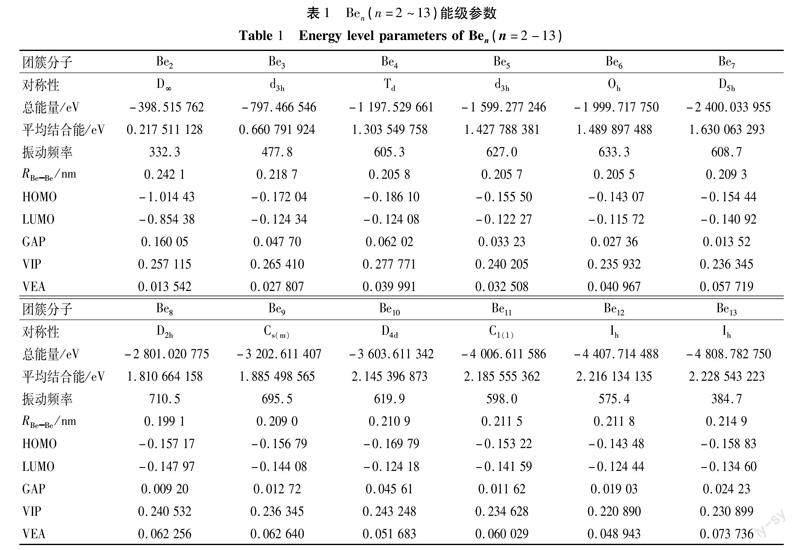
为了检测该方法的可靠性,比较了二聚体铍的相关数据。由表1可知,Be2 的键长为0.2421 nm ,这与文献[17-18]的实验结果( r =0.2470 nm )及之前报道的理论数据(0.244 nm [19]和0.245 nm [20]) 一致,表明我们选择的方法能够较准确研究 Be n 团簇对一氧化碳的吸附行为。
2 结果与讨论
2.1 Be n 团簇的几何结构优化与电子性质
采用密度泛函理论对 Be n ( n =2~13)团簇的几何结构进行了优化,获得的最低能量结构如图1所示。Be n ( n =2~13)团簇的对称性、总能 EBe 、平均原子结合能ΔEBe 、振动频率 v e 、最高已占据轨道能级 HOMO 、最低未占据轨道能级 LUMO 、能隙 HLG、垂直电离势 VIP 以及垂直亲和势 VEA 如表1所示。表1结果显示,随着团簇 Be 原子数的增加,Be n 团簇的总能量降低,平均结合能增加,团簇越发稳定。此外,我们知道 HOMO 和 LUMO 的差值 HLG 反应的是团簇的电子结构稳定性,HLG 小,电子就容易从 HOMO 跃迁到 LUMO , 电子结构的稳定性就比较低,反之,则电子就不容易从 HOMO 跃迁到 LUMO , 电子结构的稳定性就比较高。由表1可知,Be2 和 Be4 的 HLG 较高,电子不易跃迁,结构稳定性较高,而 Be8和 Be11 的 HLG 小于0.01 , 结构不稳定。
2.2 Be n CO( n =2~13)的基态结构
为了得到 Be n CO( n =2~13)團簇的最低能量结构,模拟了两种可能的情况:一种是 CO 平行吸附在 Be n 团簇上;第二种是以 CO 分子的 C 端或者 O 端吸附在 Be n 团簇上。在对各种可能的情况进行 GGA 结构优化及频率分析后,得到的 Be n CO ( n =2~13)团簇的最低能量结构如图2所示,Be n CO ( n =2~13)基态团簇的对称性、总能量Esystem、吸附能 Eads 、振动频率ν、C O 键长、最高已占据轨道能级 HOMO 、最低未占据轨道能级 LUMO 、能隙 HLG、垂直电离势 VIP 及垂直亲和势 VEA 如表2所示;图3给出了 Be n 团簇及 Be n CO 团簇中 Be Be平均键长;图4为 Be n CO 团簇中 C , O 原子间距。
Be2 CO 的基态几何结构是具有 C s ( m )对称性的四边形,是体系中仅有的平面结构,相比于纯 Be2 团簇,吸附使得 Be Be的平均键长缩短了13.71% , 而 C O 键长则伸长了13.23%;Be3 CO 的基态是具有 C s ( m )对称性的六面体结构,这是 Be n CO 体系中的第一个三维结构,此时 Be Be的平均键长缩短了8.28% , 而 C O 键长则伸长了36.46%;Be4 CO 仍是具有 C s ( m )对称性的空间结构。值得注意的是,从图3可以看出,n =4开始,吸附后的 Be Be平均键长都高于吸附前的 Be Be平均键长,此时 C , O 原子间距仍被拉长;Be5 CO 的基态结构是在 Be4 CO 的基础上添加一个铍原子得到的,具有 C 1对称性,此时 C O 键长变为0.1436 nm , 伸长了25.86% , 而铍原子平均键长只缩短了5.12%;Be6是正八面体结构,具有较高的对称性,吸附后对称性降为 C 1 , C O 键长也发生了较为明显的伸长,相比于 C 端吸附以及 O 端吸附,此时能量为-1999.7 eV , 是几种可能的结构中最稳定的结构。Be7 CO 是具有 C 1 对称性的空间结构,CO 分子平行吸附于铍团簇的表面,吸附后 C O 键长伸长了0.0246 nm 。Be8 团簇可以看作具有 D2h 对称性的双帽三角反棱柱,CO 分子的吸附使团簇的对称性降为 C s ( m ) , 同时减弱了 C O 与 Be Be之间的相互作用。从 Be9 CO 到 Be13 CO , 每种结构都可以由前一种结构加上额外的 Be 原子产生。Be9 CO 可以看作是在 Be8 CO 的边上添加了一个 Be 原子,此时 Be9 CO 中 C O 键长为0.1518 nm , 相比吸附前伸长了0.0377 nm , 说明吸附明显减弱了 CO 分子间的相互作用。Be10 CO 则可以看作是在 Be9 CO 的顶端四面体底部添加了一个 Be 原子, Be10 CO 具有与 Be9 CO 相同的 C 1 对称性。Be10 CO 端四棱锥底部连接一个 Be 原子形成 Be11 CO 最低能量结构,Be11 CO 仍具有 C 1 对称性,但是 C O 键的伸长量显著小于相邻团簇的伸长量。以此类推, Be12 CO 的最稳定结构可以通过将 Be 原子附着到 Be11 CO 的底部来获得,此时团簇对称性由原来的Ih降为 C 1 , 这表明 CO 分子的吸附极大地降低了 Be12 团簇的对称性。Be13 CO 的基态结构则是在 Be12 CO 的中心添加一个 Be 原子得到的,与 Be12 CO 相同,此时的对称性也降到了 C 1。通过图4可以看出 Be3 CO , Be6 CO , Be9 CO , Be13 CO 的 C O 键长相对较长,而 Be2 CO , Be7 CO , Be11 CO 的 C O 键长较短,这表明在 n =3 , 6 , 9 , 13时 Be n 团簇对一氧化碳分子的吸附强度高于 n =2 , 7 , 11时的吸附强度。
由上述分析可得:(1) CO 分子倾向于平行吸附在 Be n 团簇的表面并占据铍团簇的外部位置,且吸附后主团簇结构没有发生较大突变。(2) CO 的吸附会使 Be n 团簇的对称性下降。(3) CO 分子的吸附会使 Be Be的平均键长、C O 键长发生不同程度的伸长,被吸附后活性提高。
2.3 能量性质和相对稳定性
团簇的结合能是团簇热力学稳定性的度量。从表2和图5可以发现,所有 Be n CO 团簇的结合能都大于相应的纯 Be n 团簇的结合能,并且随着铍原子数的增加,纯铍团簇的结合能逐渐增加,在 n =13时达到最大值2.229 eV 。同时,Be n CO 团簇的结合能逐渐减小,在 n =7时达到最小值2.826 eV , 然后在2.900 eV 附近振荡。这表明所有 Be n CO 团簇在能量上都比相应的铍团簇更稳定。从表2和图5中 Be n 团簇与一氧化碳之间的吸附曲线可以看出,吸附能在 n =3 , 6 , 8时出现了峰值,这表明此时铍团簇和一氧化碳的相互作用较强,吸附后的结构较为稳定,且在 n =8之后团簇的吸附能出现了持续下降,即 Be8 CO 团簇相对于其他团簇更稳定;在 n =4 , 11时出现了最小值,表明 Be4 CO , Be11 CO 团簇的稳定性较低。
此外,垂直电离势( VIP)常被用来研究小团簇的化学稳定性,VIP 越大,HOMO 能级越深,反应活性越低,化学稳定性越高。 HOMO - LUMO 能隙HLG 是用于考查团簇电子稳定性的另一个重要参数。HLG 越大,从价带到导带激发电子所需的能量越高,对应的稳定性越高。图6分别给出了吸附前后团簇的 GAP 随团簇尺寸的变化规律。对于纯 Be n 团簇,其 HLG 的峰值出现在 n =2 , 4 , 10 , 这与已有的研究成果是一致的。与吸附前相比,吸附 CO 后团簇的 HLG 出现了3种情况:n =5 , 6 , 8时,吸附CO 后团簇的吸附能显著提升,特别是 Be8 CO , 其 GAP 为0.058 eV , 相比于吸附前提升了0.05 eV , 表明这3个团簇具有高的热力学稳定性;n =2 , 4 , 10时,吸附后团簇的 HLG 显著减小,尤其是 Be2 CO , 此时 HLG 减小了0.04 eV , 约为吸附前的一半,这说明 CO 分子的吸附使得 Be2 CO 团簇的化学活性得到了显著提升;其他情况下,吸附前后的 HLG 变化比较小。图7给出了吸附前后团簇的 VIP 随团簇尺寸的变化关系,当 n =2 , 5 , 6 , 7 , 8 , 12时,CO 的吸附显著提升了团簇的 VIP;此外,在 Be6 CO 和 Be8 CO 处出现的小峰值以及随后的下降表明这两个团簇比它们相邻的团簇更不容易失去电子。计算了 Be n CO 的垂直电子亲合势 VEA , 结果如图8所示。图8表明,除了 CO 分子的吸附极大地降低了 Be8 的 VEA 值之外,两条曲线的趋势几乎相同。此外,在 n =6 , 8时 VEA 出现了两个极小值,这表明 Be6 CO 和 Be8 CO 团簇在获得电子方面的相对稳定性。综上所述,可得出 Be8 CO 是吸附后体系中最稳定的结构。
3 结论
采用密度泛函理论和广义梯度近似方法在 Per- dew - Wang 91水平上对 Be n CO( n =2~13)团簇进行了全电子计算,结论如下:(1) CO 分子倾向于平行吸附在纯 Be n 团簇的表面并占据铍团簇的外部位置,吸附后主团簇结构没有发生较大的突变,但是 CO 的吸附会使 Be n 团簇的对称性下降。(2) CO 分子的吸附会使 Be Be的平均键长、C O 键长发生不同程度的伸长,CO 的化学活性显著提升。(3) Be n CO 的结合能高于纯 Be n 团簇,即 Be n CO 能量稳定性高于纯 Be n 团簇,在 n =3 , 6 , 8时 Be n CO 团簇的吸附能最大。当 n =5 , 6 , 8时 Be n CO 团簇的能隙相比于纯 Be n 团簇有显著提升;n =2 , 5 , 6 , 8和12时,CO 的吸附显著提升了团簇的垂直电离势; n =6 , 8时出现了垂直亲和势的两个极小值。Be8 CO 是吸附后该体系最稳定的结构。
参考文献
[1] LI J Y , WU D , LI Y , et al. A comparative study of oxygen- doped and pure beryllium clusters based on structural ,energetic and electronic properties [J]. Chemical Physics Letters , 2017 , 674:1-5
[2] HEAVEN M C , MERRITT J M , BONDYBEY V E . Bond-ing in beryllium clusters [ J]. Annual Review of Physical Chemistry , 2011 , 62:375-393.
[3] MARTIN J M L. The ground-state spectroscopic constantsof Be2 revisited [ J]. Chemical Physics Letters , 1999 , 303(3/4):399-407.
[4] ST?RCK J , MEYER W. The ground state potential of theberyllium dimer [ J ]. Chemical Physics Letters , 1996 , 258(3):421-426.
[5] 張文献,刘磊,李郁芬.铍团簇 Be n ( n =2-55)的基态能量、结构及其拓扑性质[ J].原子与分子物理学报,1999 , 16(4) , 592-601
[6] WANG J L , WANG G H , ZHAO J J. Density functional study of beryllium clusters , with gradient correction [ J].Journal of Physics : C ondensed Matter , 2001 , 13: L753- L758.
[7] KAWAI R , WEARE J H . From van der Waals to metal-lic bonding: the growth of Be clusters [ J]. Physical Re- view Letters , 1990 , 65(1/2):80-83.
[8] KNIGHT W D , CLEMENGER K , DE HEER W A , etal. Electronic shell structure and abundances of sodiumclusters [ J]. Physical Review Letters , 1984 , 52(24):2141-2143.
[9] EKARDT W. Work function of small metal particles : self-consistent spherical jellium-background model[J]. Physi- cal Review B , 1984 , 29(4):1558-1564.
[10]葛桂贤,井群,杨致,等.第一性原理对NaBe n ( n =1~12)团簇最低能量结构及其电子性质的研究[ J].物理学报,2006(9):4548-4552.
[11] GE G X , YAN Y L , REN F Z , et al. Density functionaltheory study of structure and electronic properties of Mg- Be n ( n =2-12) clusters [J]. Chinese Journal of Chemi- calPhysics , 2007 , 20(5):518-524.
[12]葛桂贤.过渡金属 M(M = Fe , C o , Ni)掺杂纯铍团簇的第一性原理研究[D].郑州:河南大学,2007.
[13] LEI X L , ZHAO W J , GE G X , et al. Density-functional theory study of structural and electronic properties of and clusters [ J ]. Physica B: C ondensedMatter , 2008 , 403(4):653-659.
[14] FAN H W , YANG J C , LU W , et al. Structures and electronic properties of beryllium atom encapsulated in Si n (0 , -1)( n =2-10) Clusters [ J]. Journal of Physical Chemistry A , 2010 , 114 , 1218-1223.
[15]李淑萍,孟江,王继刚. NO 在金属 Be n ( n =2~12)团簇表面的平行吸附[J].原子与分子物理学报,2019 , 36(2):240-245.
[16]董兰,蒋树斌,郑申声,等. Be n ( n =1~6)团簇吸附H2的密度泛函研究[ J]. 原子能科学技术,2010 ,44(12):1414-1419.
[17] RUETTE F , S?NCHEZ M , A EZ R , et al. Diatomic molecule data for parametric methods . I [ J]. Journal ofMolecularStructure : THEOCHEM , 2005 , 729(1/2):19-37.
[18] BONDYBEY V E . Electronic structure and bonding ofBe2[ J ]. Chemical Physics Letters , 1984 , 109(5):436-441.
[19] PATKOWSKI K , SPIRKO V , SZALEWICZ K. On theelusive twelfth vibrational state of beryllium dimer [ J]. Science , 2009 , 326(5958):1382-1384.
[20] MERRITT J M , BONDYBEY V E , HEAVEN M C .Beryllium dimer : caught in the act of bonding[ J]. Sci- ence , 2009 , 324(5934):1548-1551.
猜你喜欢
小星星·阅读100分(低年级)(2023年9期)2023-10-28
物理学报(2021年12期)2021-07-01
数学物理学报(2020年6期)2021-01-14
青岛大学学报(工程技术版)(2019年2期)2019-09-10
陕西理工大学学报(自然科学版)(2015年4期)2016-01-16
中学化学(2015年8期)2015-12-29
都市家教·下半月(2014年5期)2014-08-07
压缩机技术(2014年3期)2014-02-28
华南师范大学学报(自然科学版)(2013年1期)2013-10-27
火炸药学报(2012年4期)2012-01-29
