
基于指纹图谱与化学计量学的藿香正气软胶囊质量控制方法研究△
2024-04-12 10:05:04马宿杉苏建李舒琪苏紫藤王常顺刘永利
中国现代中药 2024年2期
马宿杉,苏建,李舒琪,苏紫藤,王常顺*,刘永利*
1.河北科技大学,河北 石家庄 050000;2.河北省药品医疗器械检验研究院/河北省中药质量评价与标准研究重点实验室,河北 石家庄 050000;3.河北中医学院,河北 石家庄 050000
藿香正气软胶囊源于宋代的《太平惠民和剂局方》,首先由宋代医者制成散剂服用,后又将散剂制成了丸剂,其由苍术、陈皮、厚朴(姜制)、白芷、茯苓、大腹皮、生半夏、甘草浸膏、广藿香油和紫苏叶油组成,具有解表化湿、理气和中的功效[1-2],用于外感风寒、内伤湿滞或夏伤暑湿所致的感冒[3]。中药指纹图谱是中药谱效关系研究的基础,能够实现对中药内在质量的综合评价和整体物质的全面控制[4]。藿香正气软胶囊现行质量标准为《中华人民共和国药典》(以下简称《中国药典》)2020年版(一部)[1],要求对苍术、陈皮、厚朴、白芷、甘草和广藿香进行薄层鉴别,对陈皮、厚朴进行含量测定。该方法质量控制项目较完善,但方法繁琐。现有藿香正气软胶囊的研究主要集中在含量分析方面[5-8],未见指纹图谱相关研究报道。因此,本研究采用高效液相色谱法(HPLC)对其中10 个指标成分进行含量测定,建立指纹图谱,结合化学计量学方法进行全面分析,进一步解析藿香正气软胶囊的质量特性,为建立更加完善的质量控制方法提供参考。
1 材料
1.1 仪器
Ultimate 3000 型高效液相色谱仪(美国Thermo公司);XPE26 型、XS105DU 型分析天平(瑞士梅特勒-托利多公司);Milli-Q 型超纯水仪(美国Millipore公司)。
1.2 试药
对照品甘草苷(批号:111610-201106,纯度:93.7%)、柚皮芸香苷(批号:112080-202201,纯度:100.0%)、橙皮苷(批号:110721-201316,纯度:95.3%)、新橙皮苷(批号:111857-201102,纯度:99.6%)、和厚朴酚(批号:110730-201313,纯度:99.1%)、厚朴酚(批号:110729-201714,纯度:100.0%)、欧前胡素(批号:110826-201918,纯度:99.0%)、异欧前胡素(批号:110827-202113,纯度:100.0%)、苍术素(批号:111924-201102,纯度:99.5%)均购于中国食品药品检定研究院;对照品花椒毒酚(批号:2009-24-7,纯度:98.0%)、白当归素(批号:482-25-7,纯度:98.0%)、水合氧化前胡素(批号:2643-85-8,纯度:98.0%)、白当归脑(批号:26091-79-2,纯度:98.0%)、珊瑚菜素(批号:2543-94-4,纯度:98.0%)均购于上海诗丹德标准技术服务有限公司;对照品花椒毒素(批号:DST210407-062,纯度:98.0%)、佛手柑内酯(批号:DST210409-012,纯度:98.0%)均购于成都德思特生物技术有限公司;色谱纯乙腈[批号:F22M2A201,赛默飞世尔科技(中国)有限公司];其他试剂均为分析纯;水为超纯水。藿香正气软胶囊样品共15 批,来自A 企业12 批(批号分别为22053111、20102211、21010911、22022811、21031611、20020411、20070111、21041611、20112111、22021711、20042211、22031912)、B企业3批(批号为2930160~2930162)。
2 方法与结果
2.1 指纹图谱和含量测定条件
以十八烷基硅烷键合硅胶为填充剂,以乙腈(A)-0.1%磷酸水溶液(B)为流动相,梯度洗脱(0~15 min,10%~47%A;15~20 min,47%A;20~30 min,47%~60%A;30~40 min,60%A;40~41 min,60%~10%A;41~50 min,10%A);流速为1.0 mL·min–1;检测波长为254、283 nm;柱温为30 ℃。
2.2 溶液的制备
2.2.1 对照品溶液的制备 取各对照品适量,精密称定,加甲醇制成甘草苷、柚皮芸香苷、柚皮苷、橙皮苷、水合氧化前胡素、花椒毒素、白当归脑、欧前胡素、和厚朴酚、珊瑚菜素、异欧前胡素、厚朴酚质量浓度为2、200、23、27、12、8、12、24、13、8、18、20 μg·mL–1的混合溶液,即得。
2.2.2 供试品溶液的制备 取本品内容物约0.2 g,精密称定,置具塞锥形瓶中,精密加入甲醇25 mL,称定质量,超声处理(功率:250 W,频率:33 kHz)30 min,放冷,再称定质量,用甲醇补足减失的质量,摇匀,滤过,取续滤液,即得。
2.3 指纹图谱
2.3.1 精密度试验 取藿香正气软胶囊(批号:20042211),按2.2.2 项下方法制备供试品溶液,按2.1 项下色谱条件,连续进样6 次,以欧前胡素峰为参照峰(S 峰),共有峰与S 峰的相对保留时间和相对峰面积的RSD 均小于1.0%,表明仪器精密度良好。
2.3.2 稳定性试验 取藿香正气软胶囊(批号:20042211),按2.2.2 项下方法制备供试品溶液,按2.1项下色谱条件,分别于0、2、4、6、8、12、24 h进样测定,以欧前胡素峰为S峰,共有峰与S峰的相对保留时间和相对峰面积的RSD 均小于1.0%,表明供试品溶液在24 h 内稳定性良好。
2.3.3 重复性试验 取藿香正气软胶囊(批号:20042211),按2.2.2 项下方法平行制备6 份供试品溶液,按2.1 项下色谱条件,以欧前胡素峰为S峰,共有峰与S 峰的相对保留时间和相对峰面积的RSD 均小于1.0%,表明该方法重复性良好。
2.3.4 指纹图谱的建立及相似度评价 取15 批(S1~S15)的样品,分别按2.2.2 项下方法制备供试品溶液,按2.1 项下色谱条件测定。将15 批样品色谱图导入“中药指纹图谱相似度评价系统”(2012版)中进行数据分析,将S1 供试品色谱图作为参考图谱,采用中位数法生成对比图谱,时间窗宽度为0.1,多点校正后自动匹配生成供试品叠加图谱与对照图谱R(图1)。通过对15 批样品指纹图谱进行分析发现,图中有22 个峰为样品溶液色谱图的共有色谱峰,由于17 号峰与相邻色谱峰分离较好、峰面积较大、tR较为稳定,因此选择17号峰(欧前胡素峰)为参照峰(图2)。15 批藿香正气软胶囊样品的指纹图谱相似度均大于0.8,表明其质量较为稳定,化学成分具有良好的一致性。
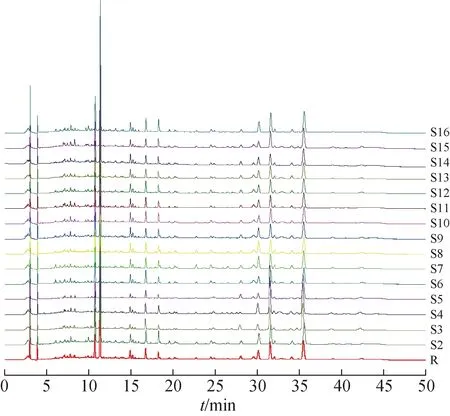
图1 藿香正气软胶囊供试品叠加图谱与对照图谱R
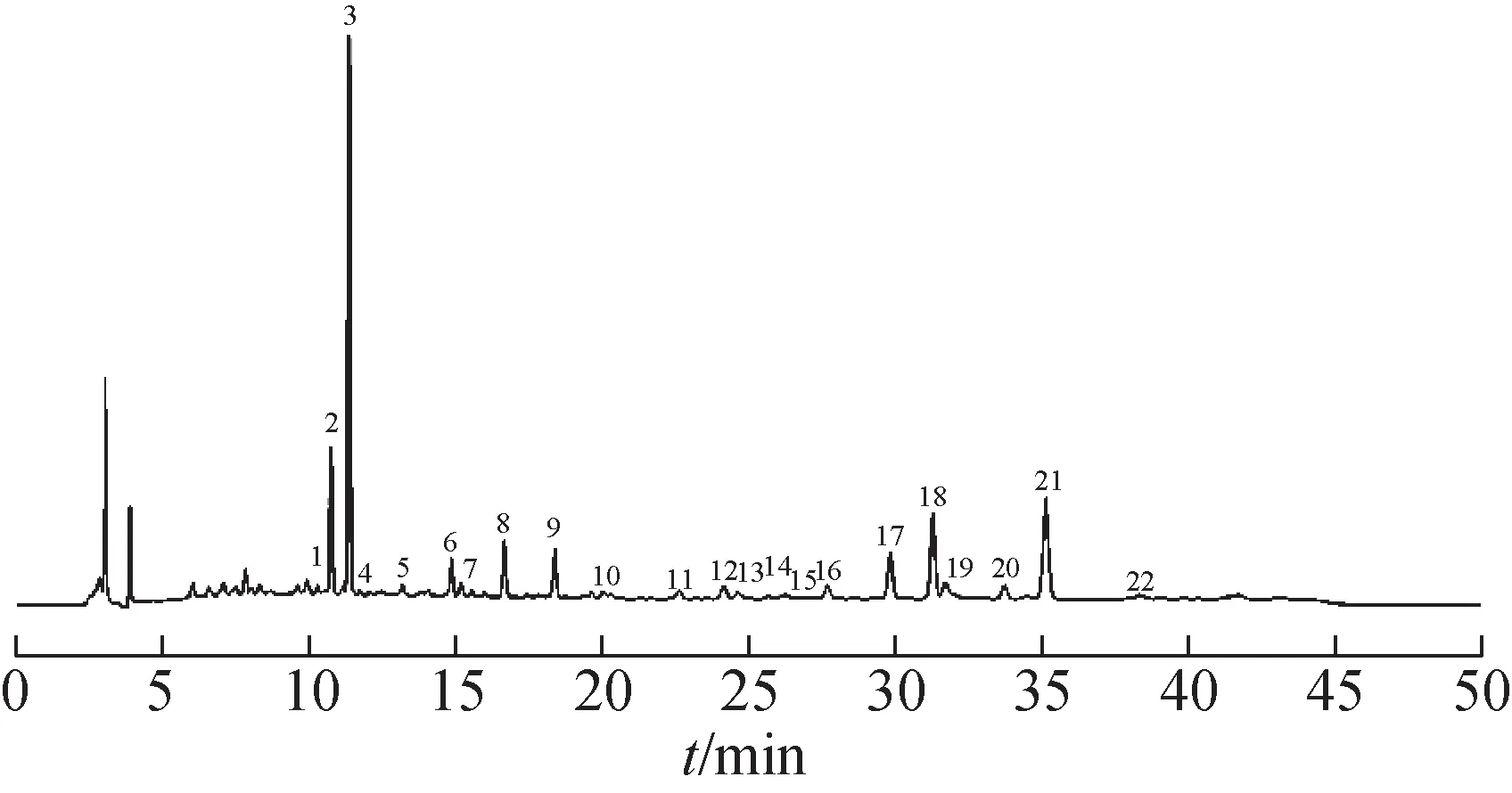
图2 藿香正气软胶囊特征图谱
2.3.5 主要色谱峰的指认 根据供试品和混合对照品的tR和紫外光谱分析结果,共指认15 个特征指纹峰(图2)。
2.4 多指标成分含量测定
2.4.1 线性关系考察 取各对照品适量,精密称定,加甲醇制成甘草苷、柚皮芸香苷、橙皮苷、水合氧化前胡素、白当归脑、欧前胡素、和厚朴酚、珊瑚菜素、异欧前胡素、厚朴酚质量浓度分别为0.3、2.0、3.0、0.2、0.2、0.5、1.0、0.2、0.4、2.0 μg·mL–1的混合溶液,摇匀,稀释6、15、30、60、150 倍,得系列混合对照品溶液,精密吸取上述系列混合对照品溶液,按2.1 项下色谱条件进样,记录色谱图(图3)。以质量浓度为横坐标(X)、峰面积为纵坐标(Y),绘制标准曲线,结果显示,各组分线性关系良好(表1)。

表1 藿香正气软胶囊中10个成分线性关系考察结果

图3 对照品和藿香正气软胶囊HPLC图
2.4.2 精密度试验 精密取同一混合对照品溶液,按2.1项下色谱条件连续进样6次,甘草苷、柚皮芸香苷、橙皮苷、水合氧化前胡素、白当归脑、欧前胡素、和厚朴酚、珊瑚菜素、异欧前胡素、厚朴酚峰面积的RSD 分别为0.50%、0.12%、0.20%、0.96%、0.41%、0.64%、1.03%、0.68%、0.63%、0.35%,表明仪器精密度良好。
2.4.3 稳定性试验 取同一供试品(批号:20042211)溶液,分别于制备后0、2、4、6、8、12、24 h,按2.1 项下色谱条件测定,甘草苷、柚皮芸香苷、橙皮苷、水合氧化前胡素、白当归脑、欧前胡素、和厚朴酚、珊瑚菜素、异欧前胡素、厚朴酚峰面积的RSD 分别为0.89%、0.80%、1.50%、0.75%、0.29%、0.62%、1.87%、0.71%、0.23%、0.20%,表明供试品溶液24 h 内稳定性良好。
2.4.4 重复性试验 采用同一批藿香正气软胶囊(批号:20042211),按2.2.2 项下方法配制6 个样品,按2.1 项下色谱条件测定其中10 个成分的质量分数,甘草苷、柚皮芸香苷、橙皮苷、水合氧化前胡素、白当归脑、欧前胡素、和厚朴酚、珊瑚菜素、异欧前胡素、厚朴酚质量分数的RSD分别为0.62%、1.45%、0.60%、0.79%、1.76%、0.66%、1.46%、1.35%、1.81%、0.85%,表明方法重复性良好。
2.4.5 加样回收率试验 取同一批藿香正气软胶囊(批号:20042211)内容物6 份,每份0.1 g,精密称定,分别加入对照品溶液,计算回收率,如表2所示,方法的重复性良好。

表2 藿香正气软胶囊中10个成分加样回收率试验结果
2.4.6 样品含量测定 将15批样品按2.2.2项下方法配制供试品溶液,按2.1 项下条件进样,采用外标法测定10 个成分的质量分数(表3)。
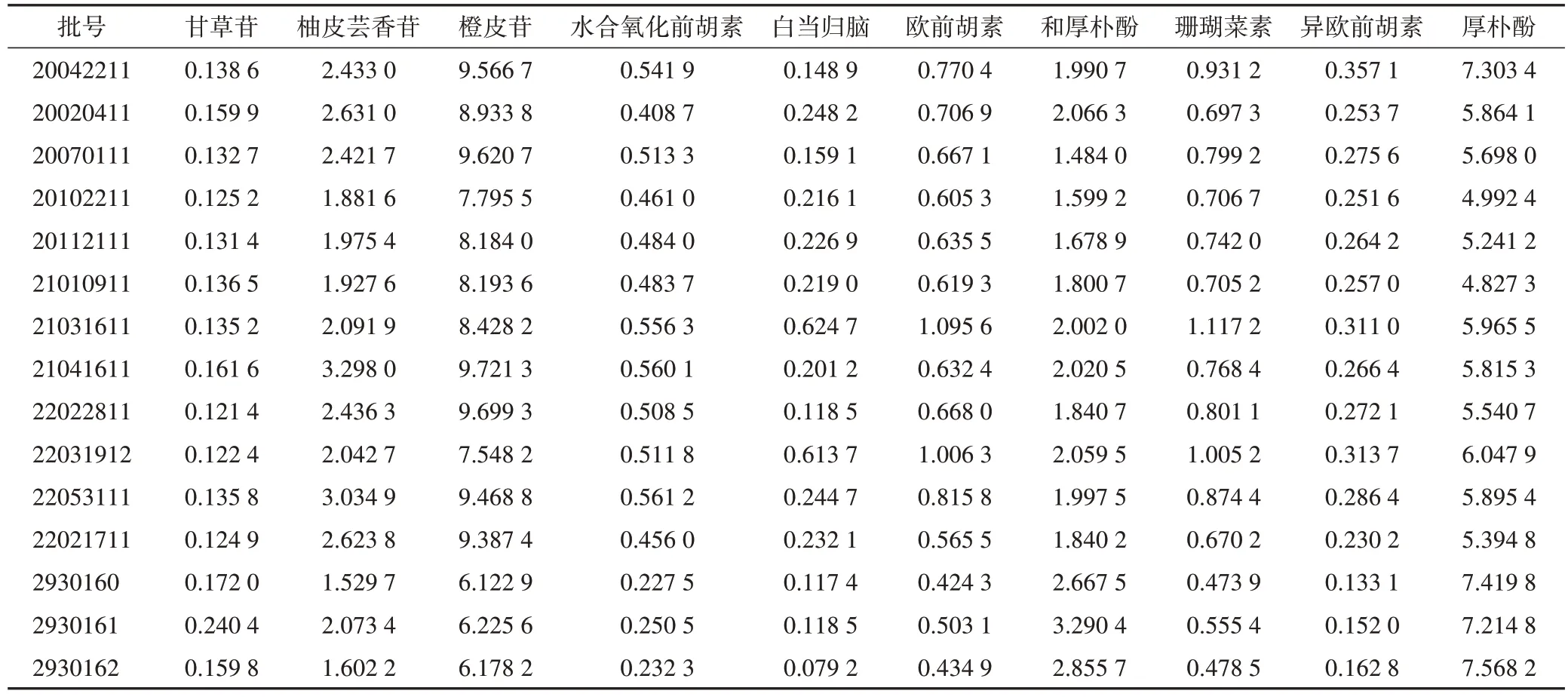
表3 15批藿香正气软胶囊中10个成分的质量分数 mg·g–1
不同企业藿香正气软胶囊10 个成分质量分数的箱线图(图4)显示,B 企业的甘草苷、和厚朴酚、厚朴酚质量分数比A 企业高,其余7 个成分A 企业普遍较高,进一步说明不同企业的不同制备工艺对藿香正气软胶囊中指标成分的含量有影响。

图4 不同企业藿香正气软胶囊10个成分的质量分数的箱线图(n=15)
2.5 化学模式识别
2.5.1 聚类分析 采用SPSS 26.0 软件,以共有峰峰面积为变量,对15 批样品进行聚类分析,结果见图5。根据树状图可以看出,15 批藿香正气软胶囊样品被分为2 个类群。相同生产厂家的样品聚为一类,说明同一企业不同批次的产品具有较高相似度,但不同企业的样品存在差异。
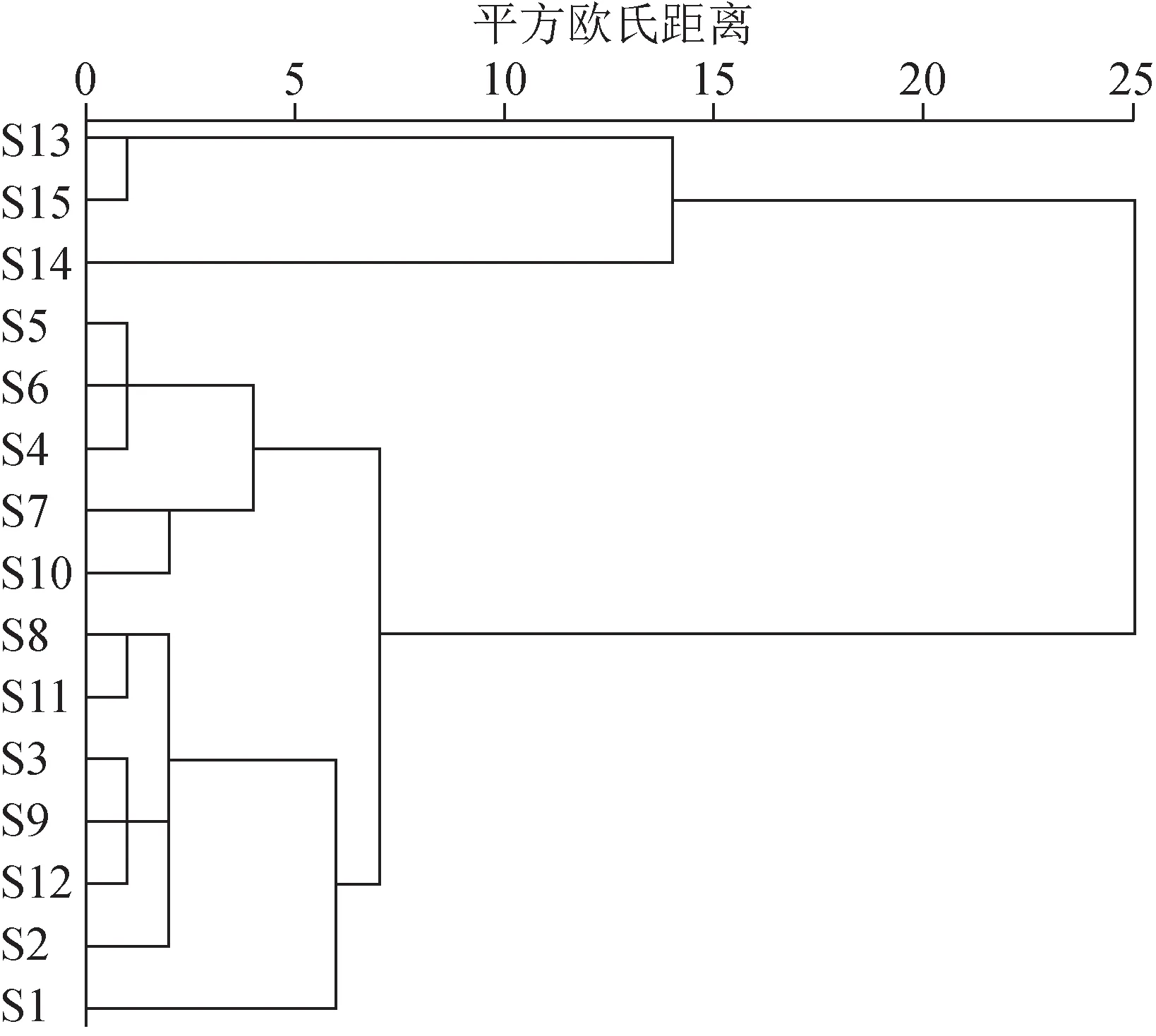
图5 15批藿香正气软胶囊的聚类分析
2.5.2 主成分分析 将15 批样品的共有峰峰面积数据导入SPSS 26.0 软件,判断的依据是特征值与累积方差贡献率,计算主成分的特征值与方差贡献率,从而对主成分进行提取[9]。以特征值>1 为提取标准,可得前2 个主成分的特征值分别为6.336 和1.975,方差贡献率分别为63.362%和19.750%,累积方差贡献率为83.112%,可以反映藿香正气软胶囊的综合质量,故选取前2 个主成分进行评价。同时,采用SIMCA 14.1 软件以10 个成分的含量数据构建主成分分析模型(图6),提取出2 个主成分的自变量拟合指数(R2X)为0.967,表明所建立的模型稳定性较高。

图6 15批藿香正气软胶囊的主成分分析得分
2.5.3 正交偏最小二乘法-判别分析 采用SIMCA 14.1 分析软件,以15 批样品共有峰峰面积为变量,采用正交偏最小二乘法-判别分析对15 批样品进行研究,得到了正交偏最小二乘法-判别分析模型(图7)。与变量重要性投影(VIP)值相结合,筛选15 批样品的主要共有峰。以VIP 值>1 为标准,筛选出6 个差异性标志物的色谱峰,分别为6 号峰(水合氧化前胡素)、18 号峰(和厚朴酚)、20 号峰(异欧前胡素)、19 号峰(珊瑚菜素)、21 号峰(厚朴酚)和1 号峰(甘草苷),上述成分对分类的影响较大(图8)。
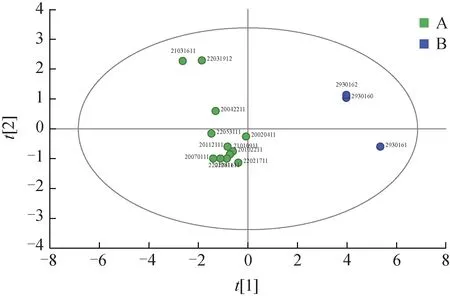
图7 不同企业藿香正气软胶囊的正交偏最小二乘法-判别分析
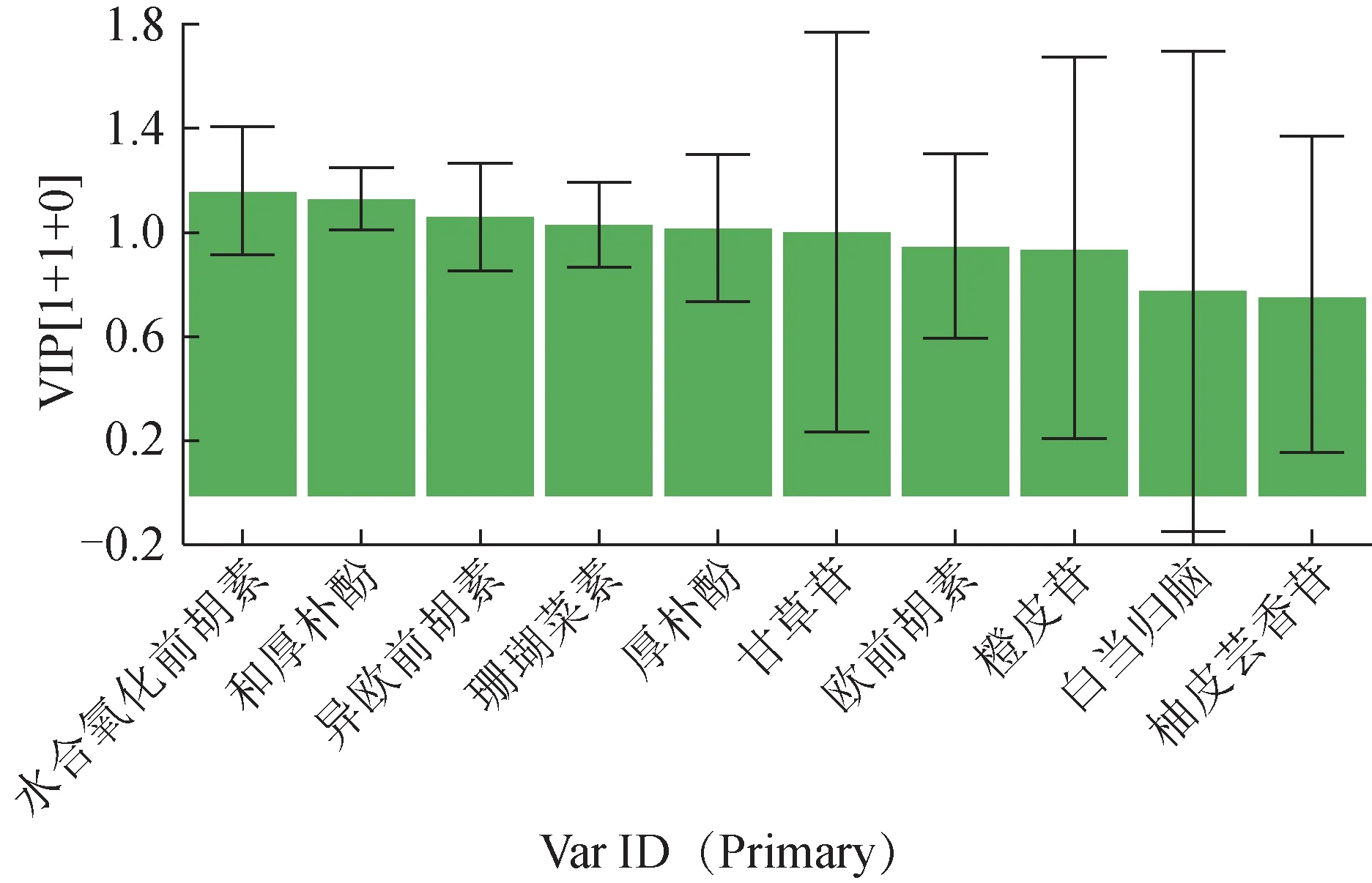
图8 15批藿香正气软胶囊10个共有峰的VIP值(±s,n=15)
3 讨论
3.1 色谱条件的选择
在前期研究中,通过改变流动相系统(乙腈-水、乙腈-0.1%磷酸水溶液等)、柱温(28、30、32 ℃),对色谱条件进行考察,结果显示,当流动相为乙腈-0.1%磷酸水溶液(梯度洗脱)、柱温为30 ℃、流速为1 mL·min–1时所得色谱图的基线平稳、色谱峰的分离效果较好。分析《中国药典》2020 年版中白芷、陈皮、厚朴、北苍术、茅苍术药材含量测定项下要求发现,本研究中22 个成分的检测波长有较大差异,因此选择采用梯度波长(254、283 nm)整合不同tR最佳色谱信息,根据《中国药典》2020 年版要求及前期实验结果确定采用2.1 项下色谱条件。
3.2 耐用性考察
本研究考察了不同色谱柱[COSMOSIL 5 C18-MS-Ⅱ(250 mm×4.6 mm,5 µm)、Capcell pak C18MG Ⅱ(250 mm×4.6 mm,5 µm)及SHIMADZU VP-ODSC18(250 mm×4.6 mm,5 µm)]、不同流速(0.9、1.0、1.1 mL·min–1)、不同柱温(25、30、35 ℃)等对藿香正气软胶囊指纹图谱和10 个成分含量测定结果的影响,发现色谱条件的变化对各成分含量测定结果影响不大,说明本方法具有较好的耐用性。
3.3 结果分析
采用中药指纹图谱技术建立了15 批藿香正气软胶囊的HPLC 指纹图谱,并鉴定出22 个共有峰,通过与混合对照品色谱图进行比对,指认出15 个共有成分。对15 批样品进行相似度评价,结果表明,15批样品与标准指纹图谱相似度都在0.8 以上,说明了不同批样品在化学组成上有很好的一致性。聚类分析结果显示,15 批藿香正气软胶囊样品可分为2类,相同生产企业的样品分为一类,主成分分析结果与其一致。采用正交偏最小二乘法-判别分析筛选得到水合氧化前胡素、和厚朴酚、异欧前胡素、珊瑚菜素、厚朴酚和甘草苷6 个差异性成分,可作为评价不同批次藿香正气软胶囊的差异性标志物。
4 结论
本研究以15 批藿香正气软胶囊样品为研究对象,建立指纹图谱及多成分含量测定方法,该方法准确、简便、重复性好,能够表征藿香正气软胶囊的整体质量,可用于藿香正气软胶囊的质量控制。采用化学计量学方法对不同批次藿香正气软胶囊的质量进行分析,筛选出不同批次藿香正气软胶囊质量差异性标志成分,本研究结果可为藿香正气软胶囊的进一步研究提供参考。
[利益冲突]本文不存在任何利益冲突。
猜你喜欢
中老年保健(2022年6期)2022-11-25 13:49:39
南昌大学学报(理科版)(2021年5期)2021-12-16 08:21:16
基层中医药(2021年8期)2021-11-02 06:24:56
基层中医药(2021年10期)2021-06-05 07:15:26
安徽中医药大学学报(2019年6期)2019-12-05 02:32:08
中成药(2018年10期)2018-10-26 03:41:02
中成药(2016年8期)2016-05-17 06:08:16
药学研究(2015年11期)2015-12-19 11:10:54
质谱学报(2015年5期)2015-03-01 03:18:47
中国当代医药(2015年8期)2015-03-01 02:01:32
