
野生型非洲猪瘟病毒多重数字PCR方法的建立
2024-04-10 01:55李松达蒋亚君庞忠宝翟文竹朱鸿飞赵晓民
动物医学进展 2024年4期
李松达,蒋亚君,庞忠宝,黄 颖,翟文竹,朱鸿飞,赵晓民,贾 红*
(1.中国农业科学院北京畜牧兽医研究所,北京 100193;2.西北农林科技大学动物医学院,陕西杨凌 712100)
非洲猪瘟病毒(African swine fever virus,ASFV)是引起非洲猪瘟(African swine fever,ASF)的病原体,系非洲猪瘟病毒科、非洲猪瘟病毒属的唯一成员,也是唯一已知的DNA虫媒病毒[1]。双链DNA基因组包含多达167个ORF,其中包括68个结构蛋白和100多个非结构蛋白,不同毒株基因组的差异很大程度上是由于病毒编码的多基因家族(multigene family,MGF)的ORFs的获得或丢失造成的[2]。p72是感染猪血液中检测到的主要抗原,基于p72建立的检测方法是目前市场上最常规普及的ASFV检测技术。CD2v蛋白由EP402R基因所编码,其转录和翻译发生在病毒复制的晚期阶段,与T淋巴细胞表面黏附受体CD2同源,是一种跨膜糖蛋白,功能主要与组织趋向性、免疫逃避和增强病毒在宿主中的复制等进程相关。在易感细胞感染非洲猪瘟病毒引起的红细胞吸附现象中,CD2v发挥不可缺少的作用[3]。UK基因在20世纪初被首次报道。在体外试验中,研究人员发现删除UK不会影响病毒在巨噬细胞中的生长,却大大降低ASFV对猪的毒力,并对亲本分离物提供了强有力的保护[4]。
针对ASF,目前尚未开发出有效且能广泛应用的疫苗。研究人员尝试开发多种不同类型的ASF疫苗,包括灭活疫苗[5]、DNA疫苗[6-7]、亚单位疫苗[8]和重组病毒活载体疫苗[9]。近几年有多项研究结果表明,ASFV减毒活疫苗(live-attenuated virus vaccines,LAVs)展现了良好的保护效果[10-11]。然而,该类减毒活疫苗大规模免疫的安全性还有待考究,距离临床应用仍相距较远。因此,还需要开发区分ASFV野毒感染与疫苗接种动物的技术,以用于区分毒力返强疫苗株和野毒株。有望在快速检测ASFV感染的同时实现基因缺失疫苗株和野生株的鉴别检测,为非洲猪瘟疫苗的研究提供技术支撑。
1 材料与方法
1.1 材料
1.1.1 病毒、细胞 p3×FLAG-CMV-B646L,p3×FLAG-CMV-EP402R,PUC57-DP96R,金斯瑞生物科技股份有限公司产品; 猪繁殖与呼吸综合征病毒(Porcine reproductive and respiratory syndrome virus,PRRSV)、猪流行性腹泻(Porcine epidemic diarrhea virus,PEDV)、伪狂犬病病毒(Pseudorabies virus,PRV)、猪水疱性口炎病毒(Vesicular stomatitis virus,VSV)、猪圆环病毒(Porcine circovirus,PCV)和ASFV病毒核酸,均由由北京畜牧兽医研究所兽医兽医公共卫生安全与管理团队提供;DH5α感受态细胞,南京诺唯赞生物科技股份有限公司产品。
1.1.2 主要试剂 Probe qPCR MixHiScript Reverse Transcriptase,南京诺唯赞生物科技股份有限公司产品;AxyPrep体液病毒DNA/RNA小量制备试剂盒,AxyPrep无内毒素质粒小量试剂盒,上海百赛生物技术有限公司北京分公司产品;RNase-free,北京依珊汇通科技有限公司产品;2×Probc dPCR Super Mix (with UNG),微液滴生成芯片,微液滴生成油,微液滴检测油,北京新羿生物科技有限公司产品。
1.1.3 主要仪器 NanoDrop2000(Nano-300)杭州奥盛仪器有限公司产品;PCR仪(veriti 96),美国赛默飞公司产品;实时荧光定量PCR仪(Gentier 96E/96R),西安天隆科技有限公司产品;微滴样品制备仪(Drop Maker M1)、生物芯片分析仪(Chip Reader R1),北京新羿生物科技有限公司产品。
1.2 方法
1.2.1 引物、探针的设计与合成 用PrimerPremier 5.0软件对ASFV的B646L、EP402R、DP96R基因的保守序列进行分析设计,获得3对荧光PCR引物及3条由不同发光基团(FAM、VIC、Cy5)标记的TaqMan探针(表1),同时将设计后的引物在生工生物工程(上海)股份有限公司进行合成。

表1 上下游引物及荧光标记探针序列
1.2.2 重组质粒标准品的制备 将p3×FLAG-CMV-B646L、p3×FLAG-CMV-EP402R、PUC57-DP96R质粒转化至DH5α感受态细胞,并涂布在具有氨苄抗性的LB平板上,进行培养。12 h后挑取白色单个菌落,接种于含有氨苄抗性的LB液体培养基,在37℃、200 r/min的摇床中进行培养。将浑浊的菌液送至北京擎科生物科技有限公司进行测序,将测序正确的重组菌进行扩大培养,并提取质粒,使用NanoDrop2000测定质粒浓度,根据拷贝数计算公式得出拷贝数。p3×FLAG-CMV-B646L、p3×FLAG-CMV-EP402R、PUC57-DP96R的拷贝数分别为1.55×1010copies/μL、1.2×1010copies/μL和1.21×1010copies/μL。
1.2.3 多重 PCR 条件的优化及标准曲线的建立 以步骤1.2.2中所获得的稀释至同一数量级拷贝数(1×1010)后的3种质粒标准品进行等比例混合后,得到B646L-EP402R-DP96R质粒混合标准品,依次进行10倍稀释,稀释至100拷贝作为模板,按照反应条件使用矩阵法进行优化,摸索体系中引物浓度、探针浓度以及退火温度,在20 μL体系中分别加入Probe qPCR Mix 10 μL、DNA模板2 μL,将3种引物、探针组合使引物终浓度为10 μmol/L,分别加入0.2、0.4、0.6、0.8、1.0 μL,浓度为10 μmol/L)探针分别加入0.1、0.2、0.3、0.4、0.5 μL,退火温度为54℃、56℃、58℃、60℃、62℃,同时设以RNase-free水模板的的阴性对照。进行多重荧光定量PCR反应,根据结果的Ct值,筛选出最合适的反应条件与体系。以标准品初始拷贝数的对数为横坐标、3种质粒标准品各稀释度Ct值为纵坐标,绘制标准曲线。
1.2.4 ddPCR 按照2×Probc dPCR Super Mix (with UNG) 15 μL,10 μmol/L上、下游引物各2.4 μL,10 μmol/L探针,0.75 μL,模板DNA 2 μL,dd H2O补足30 μL。配制相应的反应液,做好标记,进行振荡离心。将微液滴生成芯片装入配套的生成微液滴的芯片机械卡具中,水孔中加入30 μL待测样本反应液,油孔中加入180 μL微液滴生成油,盖上微液滴生成芯片密封垫。生成相应样品微液滴,按照反应程序:95℃预变性10 min;94℃变性30 s,60℃退火延伸60 s,45个循环,设置PCR,进行样品微液滴的扩增。在芯片上的小油孔和大油孔中分别加入430 μL和500 μL微液滴检测油,启动生物芯片分析仪运行,进行样品微液滴检测。
1.2.5 特异性试验 将实验室留存的PRRSV、PEDV、PRV、VSV、PCV使用AxyPrep体液病毒DNA/RNA小量制备试剂盒进行核酸提取,并使用HiScript Reverse Transcriptase对PRRSV、PEDV、VSV核酸进行反转录制备cDNA。将3种引物探针分别与提取的核酸、重组质粒的标准品作为阳性对照、RNase-free水作为阴性对照,按照优化条件进行多重荧光定量PCR反应以及ddPCR反应,以评价该方法的特异性。
1.2.6 敏感性试验 使用3种混合后的质粒标准品,以各个10倍稀释后标准品为模板,RNase-free水作为阴性对照。按照优化后的反应条件进行多重荧光定量PCR反应,以104质粒进行4倍稀释7个梯度进行ddPCR反应,以评价该方法的灵敏性。
1.2.7 重复性试验 使用3种混合后稀释至107、106、105、104、103、102拷贝数稀释梯度的重组质粒为模板,进行4次多重荧光定量PCR反应,选取单一拷贝数浓度进行ddPCR反应,并将试验结果进行统计学分析,计算变异系数,评价该方法组内和组间的重复性。
1.2.8 多重ddPCR方法的应用评价 使用普通PCR检测方法、多重荧光定量PCR方法和多重ddPCR方法对14份P3实验室中攻毒后14 d采集的口鼻拭子样品进行实验室样品检测,并对两种方法进行效果评价。
2 结果
2.1 质粒标准品的制备
由金斯瑞生物科技股份有限公司合成和重组质粒进行转化至DH5α感受态细胞扩大培养后的菌液送至测序,测序结果比对一致,证明阳性重组质粒DNA制备成功。
2.2 多重荧光定量 PCR 条件的优化及标准曲线的建立
分别优化混合引物浓度、混合探针浓度和退火温度等条件,当反应总体积为20 μL,以104copies/μL浓度的混合阳性质粒为模板,当反应体系中引物浓度为600 nmol/L,探针浓度为 300 nmol/L时,反应效果最佳。
将反应体系混匀后,使用荧光定量PCR仪进行反应,经优化后的反应程序:95℃预变性5 min;95℃变性30 s、60℃退火延伸30 s,45个循环。设立阴性对照。
B646L-DP96R-EP40R 107、106、105、104、103、102这6个不同的稀释浓度为标准品,根据扩增结果显示相关系数(R2)均到达了0.99(图1)。

图1 多重荧光定量扩增曲线与标准曲线
2.3 多重荧光定量 PCR特异性试验
根据荧光曲线结果可以判定,3套引物针对5种特异性样品(PRRSV、PEDV、PRV、VSV、PCV)、以及阴阳性对照样品检测结果均为阴性。同时,阳性对照出现特异性曲线(图2),试验成立。

1.DP96R;2.EP402R;3.B646L;4.5种非特异性样品及阴性对照曲线
2.4 多重荧光定量 PCR敏感性试验
以各个稀释倍数的质粒为扩增模板进行扩增,结果显示,优化好的荧光定量扩增体系具有较高的灵敏度,最低可以检测到102拷贝,Ct值循环数>35视为阴性。阴性对照Ct均>35个循环(图3)。
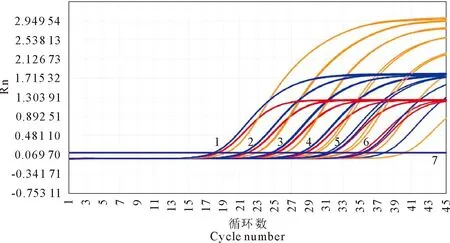
1.重组质粒浓度1×107 copies/μL;2.重组质粒浓度1×106 copies/μL;3.重组质粒浓度1×105 copies/μL;4.重组质粒浓度1×104 copies/μL;5.重组质粒浓度1×103 copies/μL;6.重组质粒浓度 1×102 copies/μL;7.重组质粒浓度1×101 copies/μL以及阴性对照
2.5 多重荧光定量 PCR重复性试验
使用107、106、105、104、103、102这6个不同的混合质粒模板浓度来检测体系中稳定性,进行4次多重荧光定量PCR反应(图4)。
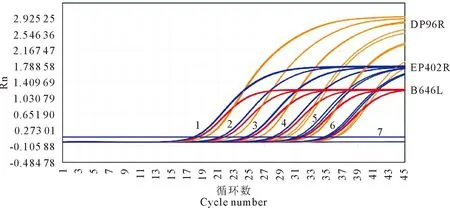
1.重组质粒浓度1×107 copies/μL;2.重组质粒浓度1×106 copies/μL;3.重组质粒浓度1×105 copies/μL;4.重组质粒浓度1×104 copies/μL;5.重组质粒浓度1×103 copies/μL;6.重组质粒浓度 1×102 copies/μL;7.阴性对照
2.6 多重ddPCR 标准曲线的建立
选择混合质粒标准品浓度为1×104copies/μL依次进行4倍稀释,获得5个不同的稀释浓度的标准品,同时设立阴性对照,ddPCR结果显示,标准曲线相关系数(R2)均大于0.999,ddPCR方法建立成功(图5)。
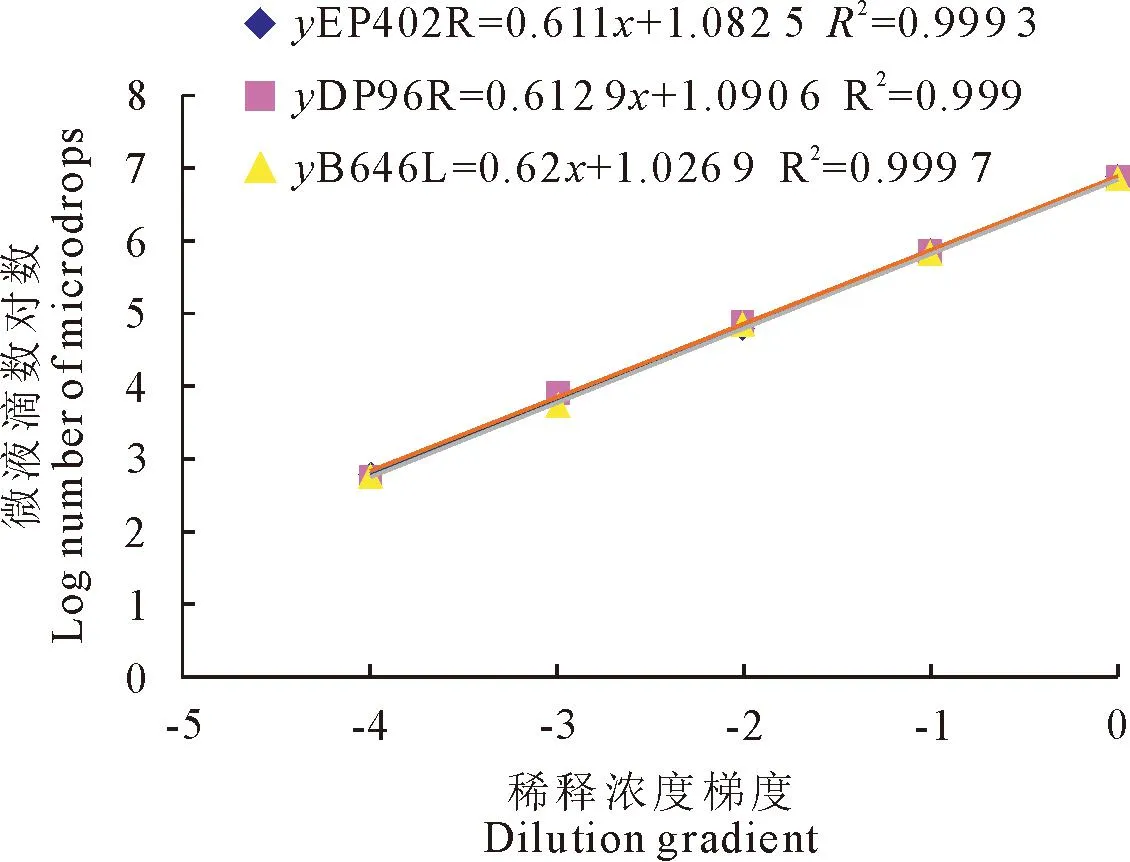
图5 多重ddPCR标准曲线
2.7 多重ddPCR特异性试验
对5种验证后的特异性样品(PRRSV、PEDV、PRV、VSV、PCV)以及阴阳性对照样品为模板进行特异性试验,结果显示,检测5种病毒及阴性对照结果均为阴性,同时阳性样品出现荧光强度,说明该方法具有良好的特异性(图6)。
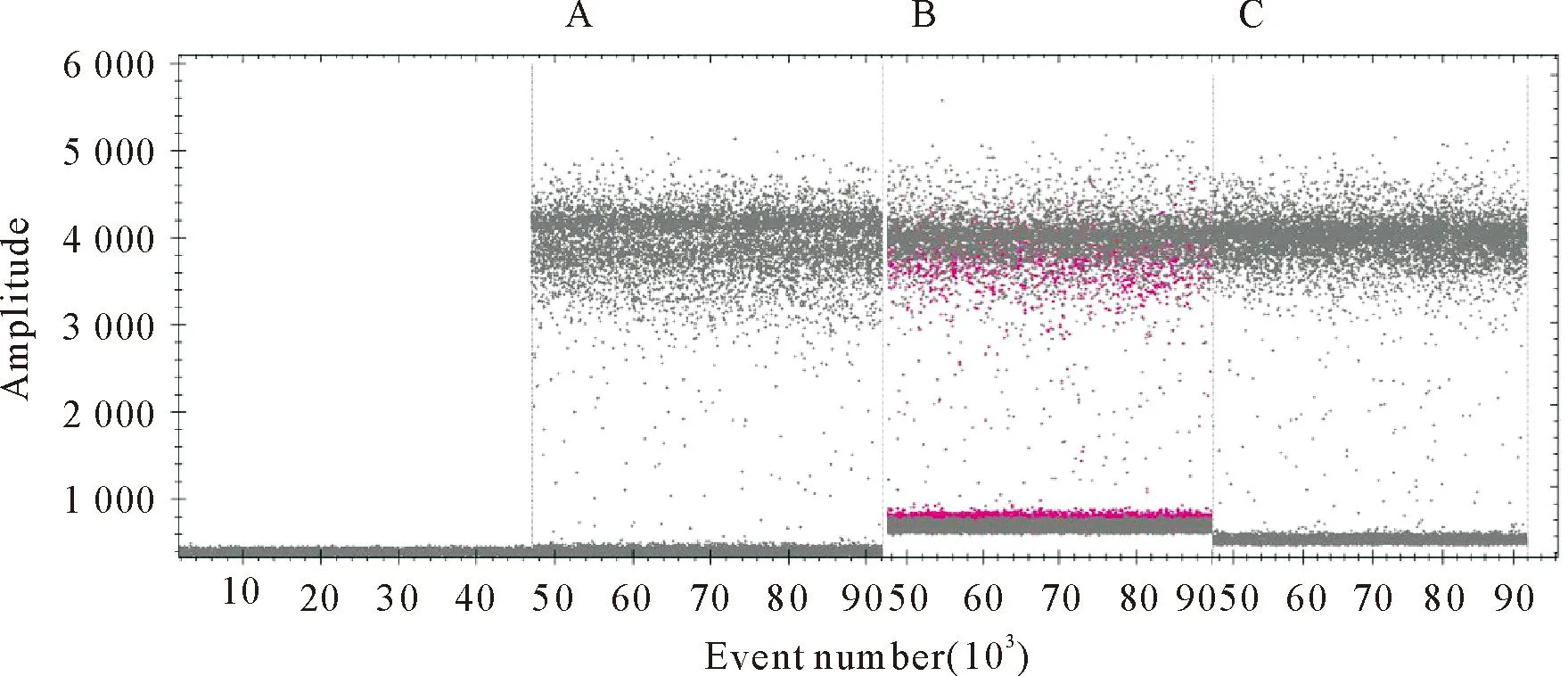
A.EP402R特异性荧光强度1D图;B.B646L特异性荧光强度1D图;C.DP96R特异性荧光强度1D图
2.8 多重ddPCR敏感性试验
选择混合质粒标准品浓度为1×104copies/μL依次进行4倍稀释,获得7个不同的稀释浓度的标准品,以此为模板进行灵敏度检测。结果表明,在三重ddPCR体系中,B646L、DP96R和EP402R检测限分别为11.6 copies/μL、16.9 copies/μL和13.3 copies/μL(图7)。
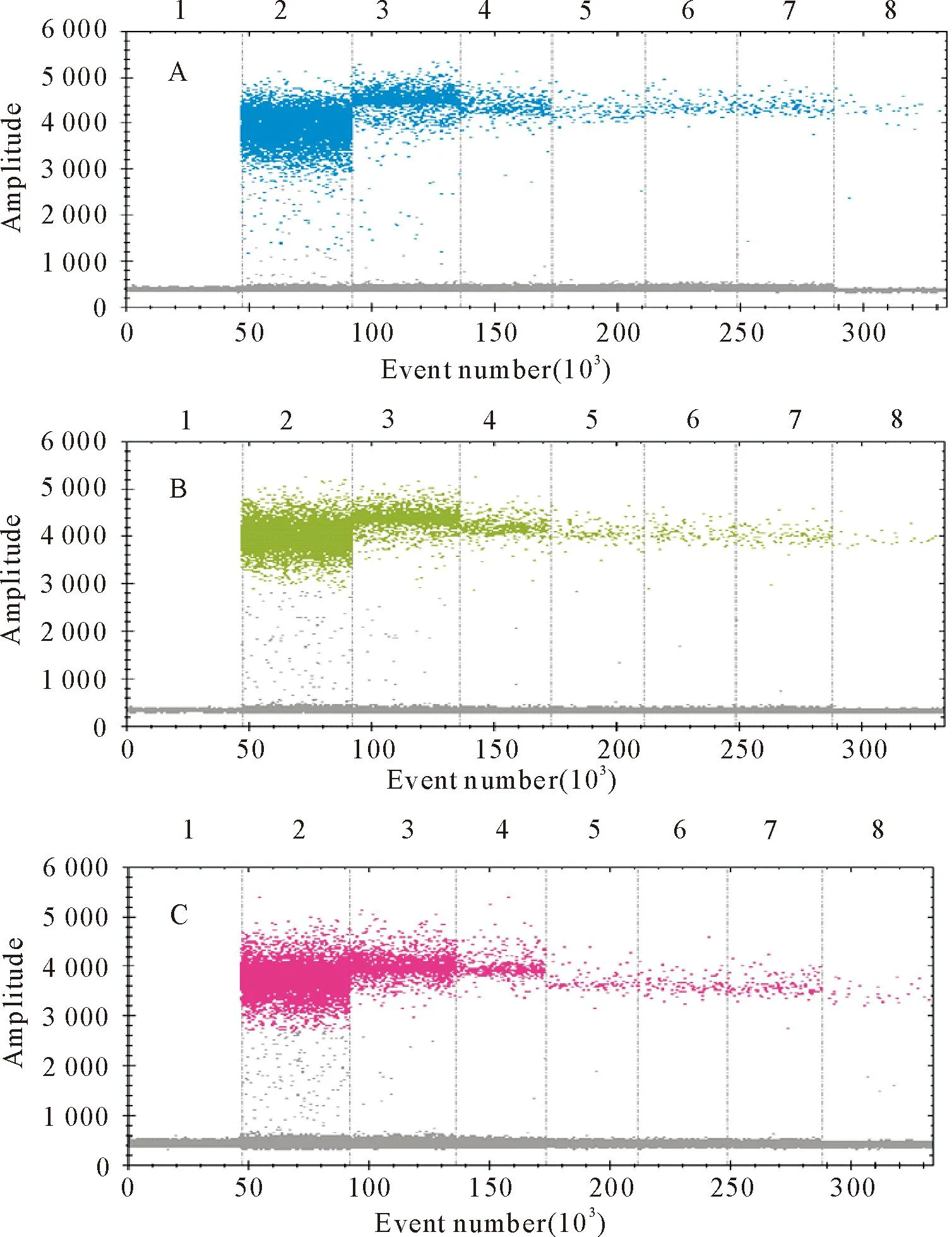
A.EP402R敏感性荧光强度1D图;B.DP96R敏感性荧光强度1D图;C.B646L 敏感性荧光强度1D图
2.9 多重ddPCR重复性试验
选取10-1次稀释后的混合质粒标准品作为阳性模板,进行组内重复实验,结果表明,组内变异系数在0.2%~0.5%之间,均小于1%,表明该方法具有良好的重复性(表2)。

表2 B646L-DP96R-EP402R重复性试验分析
2.10 多重ddPCR方法的应用评价
使用建立的多重ddPCR方法对14份P3实验室中的样品进行实验室样品检测。结果显示,样品检测符合率达到100%(表3)。

表3 临床样品检测结果
3 讨论
ASF于20世纪20年代在肯尼亚首次被描述[12-14]。2018年8月,中国记录到首例ASF疫情[15],此后该病迅速蔓延至全国各地[16]。在尚未有商品化疫苗上市时,及时发现是控制非洲猪瘟疫情蔓延的有效方法。基因缺失减毒活疫苗在近些年的研究中是开发有望的ASF疫苗,通过由单个基因或一组基因缺失组成的遗传操作获得的几种减毒株已显示诱导针对毒性亲本病毒的保护[17]。LAVs在大规模接种过程中可能存在毒力返强的风险。最后,LAVs的交叉保护能力需要进一步评估。
引物及探针设计参照文献(GenBank:MN393476.1)[18],并在NCBI查找近15年国内外流行毒株,比对B646L、EP4092R、DP96R基因的保守区序列,依据引物设计原则进行引物设计。探针分别选用FAM、VIC、CY5这3种有明显波长间隔的荧光基团,以防荧光定量PCR仪器收集荧光信号时出现的荧光相互干扰。淬灭基团分别选用BHQ1、BHQ3。FAM、VIC使用的淬灭基团为BHQ1,CY5使用的淬灭基团为BHQ3。本次试验将三重引物、探针、质粒混合,进行多重荧光定量PCR试验。
ddPCR通过极度稀释达到理论上的单分子扩增,然后利用PCR扩增和泊松分布计算样本的原始浓度[19]。同时,终点PCR信号计数检测不依赖于Ct值,不受扩增效率的影响,能够有效克服PCR抑制剂的影响。基于靶向ASFVB646L、EP402R、DP96R基因开发的多重ddPCR测定,其显示出高度的线性和特异性,并且与实时PCR相比灵敏度高了近10倍,拥有绝对定量的特性,在疫病检测诊断领域有着重大的价值。然而,ddPCR的多重荧光定量方法仍有需要改进:首先,目前市面上常用的是双荧光通道的ddPCR仪,这对于建立三重以上的ddPCR方法存在着很大的限制,建立三重ddPCR的方法也需要将探针进行双荧光标记,无疑为方法建立提高了难度。其次,ddPCR平台以及使用配套的试剂耗材的成本相对普通PCR或者TaqMan探针法荧光定量PCR的成本要高,在一定程度上也导致ddPCR方法的普及程度。最后,由于ddPCR的检测灵敏度高,以及现有常规的ddPCR检测平台为分步进行,分别是体系配置,微滴生成,PCR反应,PCR产物上机分析等几个步骤。在配置体系和转移的过程中会有污染的风险。对实验室的要求也是极高的,需要有专用体系配置的房间,加样的房间以及上机实验的房间,每个房间相对独立,尽量防止气溶胶污染影响实验。结果显示,所建立的PCR方法线性关系好。特异性良好,未与其他病原发生交叉反应。灵敏性试验得出对3种目的基因最低都可以检测到102copies/μL,重复性试验变异系数均小于2%,重复性良好。使用5荧光通道ddPCR仪,在多重荧光定量PCR基础上建立出可以同时对3种目的基因进行检测的多重ddPCR。本次试验所建立的多重ddPCR的3种引物的标准曲线R2>0.999,且重复性较好。特异性试验结果表明未与其他病原发生交叉反应,B646L可以检测出11.6 copies/μL、EP402R可以检测出13.3 copies/μL、DP96R可以检测出16.9 copies/μL,组内变异系数均小于5%,重复性良好。最后,对多重ddPCR方法进行应用评价,使用14份P3实验室中的已知样品进行实验室样品检测,样品检测符合率达到100%。成功建立了能够同时检测B646L、EP4092R、DP96R3种基因的多重荧光定量以及ddPCR检测方法,能够区分非洲猪瘟的野毒株和基因缺失株,为早期临床诊断提供了快捷高效的方法。
猜你喜欢
世界科学技术-中医药现代化(2020年2期)2020-07-25
中成药(2018年12期)2018-12-29
食品科学(2018年10期)2018-05-23
中成药(2017年6期)2017-06-13
现代检验医学杂志(2016年3期)2016-11-15
三峡大学学报(自然科学版)(2016年6期)2016-04-16
西南医科大学学报(2015年1期)2015-08-22
医学研究杂志(2015年4期)2015-06-10
中国当代医药(2015年9期)2015-03-01
物理实验(2015年9期)2015-02-28
