
先天性下肢毛细血管扩张性大理石样皮肤1例
2024-04-08 12:32刘欣欣冯小燕任敏卞亚伟李钦峰
中国皮肤性病学杂志 2024年4期
刘欣欣,冯小燕,任敏,卞亚伟,李钦峰
1 临床资料
患儿男,3个月,生后发现左下肢膝部、足部多发网状萎缩性红斑,外观呈大理石样,膝部皮损出现破溃,哭闹时暗红斑颜色加深。出生后曾于本院新生儿科住院治疗,考虑“先天性大理石样皮肤?”。患儿足月顺产,是第一胎,出生体重为2.85 kg,生后即刻Apgar10分。父母非近亲结婚,母亲孕期产检无患病及用药史,家族中无类似疾病患者。体格检查:生长发育良好,神经系统等各系统检查未见异常。皮肤科情况:左下肢、足部广泛分布网状萎缩性红斑,皮损周围可见明显毛细血管扩张,膝部红斑位置可见皮肤凹陷性瘢痕、结痂(图1)。
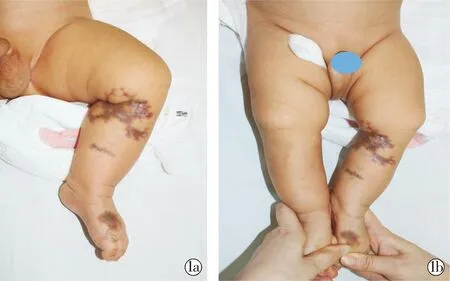
图1a~1b 左下肢及足部皮损
实验室检查:血常规、尿常规、肝肾功能、Ig+C3+C4、ANA+ENA、及梅毒、HIV血清学实验均未见异常。左下肢MRI结果示:左膝、左小腿及左足内侧皮下脂肪层内病变伴明显不均匀强化,考虑血管源性病变。膝部凹陷处皮损病理检查结果示:表皮大致正常,真皮内见弥漫增生且扩张的毛细血管及畸形静脉,管壁增厚,血管周围可见较多纤维细胞及少量炎症细胞浸润(图2)。诊断为:先天性下肢毛细血管扩张性大理石样皮肤(cutis marmorata telangiectatica congenital,CMTC)。治疗采用下肢局部皮损保暖,厚涂多磺酸黏多糖乳膏,2~3次/d,随访6个月,患儿部分皮损变淡,暂无溃疡及肢体不对称出现。
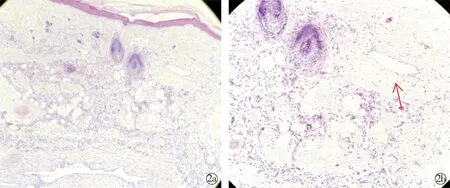
HE×100; HE×200
2 讨论
先天性毛细血管扩张性大理石样皮肤(CMTC)是一种罕见、散发的血管畸形,被国际脉管性疾病研究学会(ISSVA)归类为单纯毛细血管畸形[1]。CMTC最早于1922年由荷兰医生Cato van Lohuizen报道及命名,后续也有学者称之为网状血管性痣、先天性网状青斑、范·洛休伊泽综合征(van Lohuizen syndrome)等[2]。CMTC主要临床特征是持续性网状大理石样红斑,可能伴有溃疡或皮肤萎缩、保暖后不容易消退等特点[3]。该病可以泛发全身或局限于身体的特定区域,局部病变通常累及一侧肢体,但也可能累及躯干和面部,皮损特征性地病变不越过中线。该病病因不明,中线分离的典型节段分布表明 CMTC可能是由遗传嵌合体引起的[4]。与CMTC相关的基因突变通常是合子后体细胞的突变,最近PIK3CA、GNA11(c547C>T)、AKT3基因突变在CMTC皮损皮肤活检中相继发现,CMTC的遗传学及其相关发病机制正在继续阐明[5-6]。遗憾的是本例患儿暂未完善皮损处体细胞基因分析。目前关于CMTC诊断尚无统一标准,Kienast等[3]曾提出诊断标准,主要标准(3个)包括:先天性网状红斑、1岁内受累区域内无静脉扩张以及对局部变暖反应迟钝;次要标准(5个):2年内红斑消退、受CMTC影响的区域内出现毛细血管扩张、受 CMTC 影响的区域外出现葡萄酒色斑、溃疡和萎缩。也有研究学者建议诊断标准应增加身体不对称性。该疾病组织病理缺乏特异性,尚无统一诊断标准。目前国内报道CMTC患儿仅有3例行病理活检,结果示真皮和皮下组织毛细血管和小血管扩张。本例患儿皮损病理活检处不仅可见大量明显扩张的毛细血管并且可见畸形扩张静脉,与既往Fujita等[7]学者报道病例中的病理结果一致,因此组织病理学在CMTC的诊断中应起辅助作用,并且完善体细胞基因分析有助于该疾病发病机制探究。但病理中的血管增生对CMTC临床病程的影响尚不清楚。
由于CMTC累及毛细血管和小静脉,受累皮肤区域不仅会出现皮肤萎缩和溃疡,也可能出现身体两侧不对称。有文献回顾研究在485例CMTC患者中有37.7%观察到身体不对称,其中36.1%存在腿长差异。13.6%的腿长差异从1~6.8 cm不等,腿长差异>1 cm会导致严重的功能限制,出现骨盆倾斜、脊柱侧弯、关节疼痛等结果[2,8]。CMTC本身是一种相对良性的疾病,通常不需要治疗。皮肤病变通常会在出生后6周~26岁内消退,最常见于1岁以内,但也有不能完全消退的可能。有研究人员尝试应用脉冲染料激光和强脉冲光疗法治疗局部皮损,取得良好效果[9]。但激光治疗也增加了相关瘢痕形成的风险。该疾病往往会伴发双侧肢体不对称、先天性青光眼、神经系统发育迟缓、先天性心脏病等相关异常症状[3]。尤其青光眼和肢体不对称差异症状,如果不早起诊断和治疗,可能会产生严重后果。因此CMTC患儿在出生后应定期检查青光眼的发生,CMTC累及下肢的儿童应定期监测在儿童期直至青春期结束的腿长差异。2014年的一项研究提出腿长差异>2 cm时,应采用骨骺固定术进行治疗[10]。本例患儿病变位于左下肢,皮损尚未出现溃疡症状,部分皮损变淡,因此未采用脉冲燃料激光治疗,至目前随访近1年未出现肢体不对称症状,但仍需紧密随访至青春期结束。
猜你喜欢
世界科学技术-中医药现代化(2022年2期)2022-05-25
中国典型病例大全(2022年7期)2022-04-22
昆钢科技(2022年1期)2022-04-19
辽河(2021年12期)2021-12-24
世界科学技术-中医药现代化(2021年12期)2021-04-19
石材(2020年6期)2020-08-24
少儿美术(快乐历史地理)(2018年9期)2018-12-29
宝藏(2017年11期)2018-01-03
中华皮肤科杂志(2014年3期)2014-12-19
中国火炬(2014年8期)2014-07-24
