
植被恢复类型对露采矿山复垦土壤丰富和稀有微生物类群的影响
2024-04-08 07:34:44华子宜程彦郡朱燕峰杨永均
煤炭科学技术 2024年2期
马 静 ,华子宜 ,程彦郡 ,朱燕峰 ,杨永均 ,陈 浮
(1.河海大学 公共管理学院, 江苏 南京 211100;2.中国矿业大学 环境与测绘学院, 江苏 徐州 221116)
0 引 言
当前化石燃料主导着全球能源消费,近20 年我国煤炭开采持续扩大。煤炭开采引发的地表形变导致植被—土壤严重退化,损害矿区生态环境[1-2]。党的十八以来,中央高度重视生态文明建设,矿山环境治理与生态恢复是重中之重,备受各界关注。植被修复是矿区生态环境治理的有效方式,也是矿山生态环境恢复的重要标志。然而,植被生长严重受限于矿区复垦土壤的功能恢复,尤其是生态环境相对脆弱的黄土高原露天煤矿区。露天煤矿复垦土壤压实重、结构散、肥力差,缺少必要的碳、氮等营养元素,严重限制植被恢复的养分需求[3]。微生物是有机物分解和养分循环的重要贡献者,复垦土壤微生物区系发育直接影响土壤功能和性状,从而影响植被生长。不同的植被恢复模式又会影响土壤微生物群落组成、多样性和代谢功能等,对土壤功能和性状产生逆反馈作用[4]。因此,充分厘清土壤–植物–微生物之间的交互作用,形成一个互促互进的良性循环机制,有助于激发生态系统自恢复潜力,对黄土高原露天煤矿区生态恢复和管理至关重要。
土壤微生物种类丰富、功能复杂,不同丰度的分类群在生物地球化学循环中扮演着不同的角色。微生物群落中物种的丰度和分布是不均匀的,丰富种少,稀有种多[5]。不同的微生物在维持生态系统功能中发挥着关键作用,包括营养循环、有机质分解、土壤健康和作物生产力[6]。然而,采矿干扰直接损害微生物的正常生理代谢,进而对微生物多样性乃至生态系统产生不利影响。在土壤修复过程中,丰富微生物类群因其在生物量提高和养分循环改善中的突出贡献而被长期关注。近年来的研究开始强调稀有类群在维持生态系统稳定方面的重要性。稀有物种具有较高的多样性和功能冗余,因此在微生物群落中具有功能保障作用。研究发现,丰富和稀有物种抵抗扰动时会存在种内或种间相互作用,构成复杂的生态网络,维持微生物网络的稳定性[7]。值得注意的是,有些物种,无论其数量多少,仍在生态网络中占据关键位置(如枢纽和连接点),被认为是群落结构稳定必需的关键物种[8]。然而,无论这些关键物种是丰富或稀有种,它们对环境扰动的响应并不总是一致的[9]。例如,研究表明,丰富类群比稀有类群抵抗环境扰动方面具有更广泛的环境变化适应能力[10]。然而,由于对重金属的抗性差异,原始土壤中几乎所有的稀有类群都被重金属清除,导致细菌多样性严重下降[6]。也有研究表明,在气候变化和其他干扰(如铜胁迫、冻融和机械干扰)的影响下,稀有类群的多样性和群落组成更加稳定[8]。CHANG 等[11]发现,植被恢复增加了矿区复垦土壤微生物群落网络和复杂性,混交林网络具有更为复杂结构和稳定性。微生物共现网络方法有助于更好地理解植被恢复类型或土地利用变化对微生物群落的影响,并依据拓扑关系阐明不同关联之间的复杂性和稳定性。此外,还有研究发现在高度异质环境中丰富和稀有类群表现出复杂的相互作用,如捕食、竞争、共生等[12]。鉴于丰富和稀有微生物类群在土壤功能和生态系统服务中发挥不同作用,加上对稀有微生物的认知局限性,研究者对土壤生态系统如何响应外界扰动预测的准确性有所下降。虽然现阶段对丰富和稀有类群的关注与日俱增,但对矿区生态修复过程中复垦土壤丰富和稀有类群变化一直鲜有研究,尤其是不同植被恢复模式下丰富和稀有类群如何调节复垦土壤功能及植被—土壤之间互作关系。
植被-土壤-微生物之间交互作用和机制十分复杂,厘清三者之间关系对基于自然的矿山生态修复解决方案至关重要。为此,研究采用高通量测序技术和分子生态网络方法,分析内蒙古准格尔旗黑岱沟露天矿复垦示范基地的东排土场5 种植被恢复类型和1 个对照共48 个土样中丰富和稀有微生物群落组成、多样性及共现网络差异,探究土壤丰富和稀有微生物群落、植被和环境因素之间的互作关系及机理,揭示驱动丰富和稀有微生物群落特征差异的关键环境因子,为科学评估植被恢复如何影响矿区土壤质量动态提供依据。
1 材料与方法
1.1 研究区概况
黑岱沟露天矿东排土场(39°43′N~39°49′N,111°13′E~111°20′E)位于内蒙古自治区准格尔旗,该地区属于温带半干旱大陆性气候,年均气温7.2 ℃,年均降水量396.0 mm,集中于7—9 月份,占全年降水的70%,年蒸发量为1 824.7~2 896.1 mm。该排土场面积为210.0 hm2,为典型黄土丘陵沟壑区,主要植被为暖温型草原带,覆盖度一般小于30.0%。土壤类型为黄绵土,颜色浅、肥力低、偏碱性、比热容小。1992 年该排土场开始排弃,1997 年实施人工复绿,采用台–坡阶梯式布置,分3 层,底层海拔1 215 m,每层高差为30 m,分21 个复植区域,主要种植苜蓿、沙打旺、沙棘、锦鸡儿、紫穗槐、油松、刺槐、杨树等植被[13]。
1.2 样品采集与处理
2022 年8 月20 至25 日开展野外调查,采集对照(裸地,CK)草地(苜蓿,GL)、灌丛(沙棘,BL)、针叶林(油松,CF)、阔叶林(杨树,BF)、混交林(油松+杨树,MF),如图1 所示。通过“5 点取样法”在每块样地上采集土壤样品,即首先确定对角线的中点作为取样点,然后在对角线上选择与中心点等距的点进行取样。在取样前,详细记录当天基本天气状况、土壤含水量和温度等相关指标,同时清理样方表面腐殖质层和杂质,采集0~20 cm 表层土壤约300 g,每个植被类型采集8 个土壤样品,共计48 个。除去土壤中植物根系和凋落物残体,将土壤样品充分混合均匀,密封在聚氯乙烯袋中,写好样本编号,带回实验室,风干样品后再次剔除砂石,动植物残体等杂质,直接放入4 ℃冰箱运回实验室。土样充分混匀后,分为两份,一份用于微生物多样性和部分土壤酶活性测定。另一份在室内风干后过2 mm 筛,用于土壤理化性质和土壤酶活性的测定。土壤pH 值(水、土质量比 2.5∶1)采用电位法(PH3–C,上海雷磁),土壤有机质(SOM)采用重铬酸钾稀释热法测定,易氧化有机碳(EOOC)采用333 mmol/L-1高锰酸钾溶液氧化法测定,铵态氮(AN, NH+4-N)和硝态氮(NN,NO-3-N)采用氯化钾、氯化钙浸提–紫外分光光度法测定,有效磷(AP)釆用0.5 mol/L-1NaHCO3浸提再用钼锑抗试剂比色测定,测定步骤详见文献[14]。研究测定的土壤酶活性指标主要包括β–葡萄糖苷酶、碱性磷酸酶、脲酶、亮氨酸氨基肽酶。其中,碱性磷酸酶(ALP)通过磷酸苯二钠比色法测定,脲酶(URE)通过苯酚钠–次氯酸钠比色法测定,β–葡萄糖苷酶(BG)通过硝基苯酚比色法测定[15],亮氨酸氨基肽酶(LAP)采用索莱宝试剂盒测定。

图1 研究区位置和采样点示意Fig.1 Location of the study area and sampling sites
1.3 DNA 提取,高通量测序和微生物类群划分
土壤微生物群落多样性测定采用高通量测序法:首先,使用E.Z.N.A.® soil 试剂盒(Omega Bio-tek,Norcross, GA, U.S.)提取每个土壤样品总DNA,后采用超微量分光光度计(NanoDrop2000)经1% 琼脂糖凝胶电泳检测DNA 浓度和纯度。采用16S rDNA的V3-V4 区标准引物对338F/806R (5'-ACTCCTA CGGGAGGCAGCAG-3',5'-GGACTACHVGGGTW TCTAAT-3') 进行PCR 扩增,95 ℃预变性3 min,95 ℃ 30 s,55 ℃ 30 s,72 ℃ 45 s,27 个循环,最后72 ℃延伸10 min。通过AxyPrep DNA Gel Extraction Kit进行纯化,Tris-HCl 洗脱, 2%琼脂糖电泳检测后回收PCR 产物,再通过QuantiFluor TM-ST 定量检测。采用Illumina MiSeq (Illumina,美国) 标准方法利用纯化后的扩增片段构建测序文库,再采用美吉Miseq PE300 平台测序,该测序流程工作委托上海美吉生物医药科技有限公司完成。
研究中,门水平下,相对丰度≥1%的菌门被定义为丰富菌门,相对丰度≤0.01%的菌门被定义为稀有菌门。ASVs(Amplicon Sequence Variants)水平下,当在所有样本中ASVs 的平均相对丰度≥0.1% 时,将其定义为丰富类群;当ASVs 在整个数据集中的平均相对丰度≤0.05%时,将其定义稀有类群。相对丰度在0.05% ~ 0.1%之间的ASVs 属于中间群落[16-18]。
1.4 数据处理与统计分析
采用SPSS 26.0 软件进行数据处理与统计分析,对不同处理土壤理化、酶活性和α 多样性指数采用单因素方差分析(one-way ANOVA)进行方差齐性检验,土壤微生物群落丰度图使用OriginPro2020 软件(Origin Lab,USA)绘制,在不同处理之间的微生物群落ASVs,进行基于Bray–Curtis 距离非度量多维尺度(NMDS)分析其微生物群落β 多样性。微生物共现网络的构建及拓扑参数的计算是基于细菌和真菌高通量测序结果,采用Mena 平台进行,网络分析中,节点代表ASVs,连接节点的边代表ASVs 之间的关联性,并使用R 实现可视化。Mantel tests 利用R 中的ggcor 包进行了单因素方差分析,以确定P< 0.05处理之间的显著差异。
2 结果与分析
2.1 不同植被类型土壤理化性质和酶活性变化
排土场复垦区不同植被恢复类型对土壤产生不同影响,土壤理化性质和酶活性影响存在显著差异,如图2 所示:土壤pH 值均高于7.0,呈碱性,其中MF 最高,BL 最低且与其他处理组存在显著性差异(图2a,P< 0.05)。BL 土壤有机质含量显著高于CK、GL、BF 和MF(P< 0.05),CK 显著低于所有植被恢复组(图2b,P< 0.05)。CF 土壤易氧化有机碳含量最高,MF 最低,且二者与其他处理呈显著性差异(图2c,P< 0.05)。BL 土壤中硝态氮积累显著高于其他处理(图2d,P< 0.05)。不同植被恢复组土壤铵态氮显著高于CK,与GL、BL、CF、BF 和MF 相比,分别下降了56.5%、53.4%、54%、57.2% 和53.7%(图2e,P< 0.05)。BL 土壤中有效磷含量显著高于CK、CF、BF 和MF(P< 0.05),GL 与其他处理组差异不显著(图2f)。在土壤酶活性方面,CK 组土壤β–葡萄糖苷酶活性显著高于其他植被处理组(P<0.05),且大约提高了2.7% ~ 5.3%(图2g)。土壤脲酶、亮氨酸氨基肽酶和碱性磷酸酶活性变化趋势较为接近,其中BL 土壤中这3 种酶活性最高,显著高于其他处理组,而CK 显著低于其他处理(图2h、2i和2j,P< 0.05)。
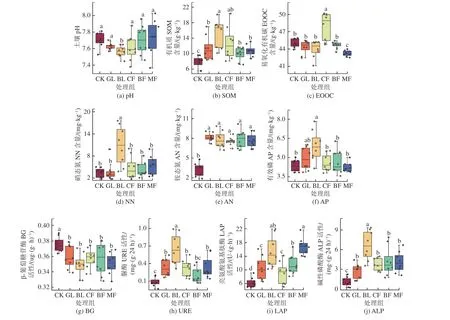
图2 不同处理组土壤理化性质以及土壤酶活性变化Fig.2 Changes of soil physicochemical properties and soil enzyme activities in different treatment groups
2.2 不同植被类型土壤丰富和稀有细菌、真菌群落结构特征
2.2.1 不同植被类型土壤丰富和稀有细菌、真菌群落组成
植被恢复类型的变化对土壤丰富和稀有细菌、真菌群落组成产生影响,有一定的差异性,但在所有处理中,土壤丰富和稀有细菌、真菌群落主要菌门保持一致。丰富细菌的优势门为放线菌门(Actinobacteria)、变形菌门(Proteobacteria)、绿弯菌门(Chloroflexi)和酸杆菌门(Acidobacteria),其中放线菌门最为丰富,其相对丰度占比在28% ~ 49%(图3a),且在CK 中占比最高,显著高于其他植被恢复组(P<0.05)。在稀有细菌类群中,装甲菌门(Armatimonadota)和蓝藻门(Cyanobacteria)为优势稀有细菌门(图3b),尽管所有处理组稀有细菌主要菌门相似,但各组相对丰度变化存在明显差异。植被恢复组蓝藻门(Cyanobacteria)的相对丰度显著低于CK(P<0.05)。GL 组的脱硫杆菌门(Desulfobacterota)丰度显著高于其他植被复垦及CK 组,而装甲菌门和蓝藻门则呈现与之相反的趋势。在丰富真菌类群中,子囊菌门(Ascomycota)和担子菌门(Basidiomycota)为群落主导菌门(图3c)。CF、BF 和MF 担子菌门显著高于其余处理组(P< 0.05)。所有处理组的稀有真菌相对丰度结果显示(图3d),BL、CF、BF 和MF 处理组的罗兹菌门(Rozellomycota)相对丰度占比最高,分别为56.1%、86.3%、88.5% 和91.63%,显著高于CK 和GL 组(P< 0.05)。CK 处理的油壶菌门(Olpidiomycota)显著高于植物恢复组,而GL 组的捕虫霉门(Zoopagomycota)和蛙粪霉门(Basidiobolomycota)则显著高于其他处理组(P< 0.05)。总体来看,随着植被恢复类型的改变,丰富和稀有细菌种类多于真菌,但真菌丰度变化更为显著,尤其是稀有真菌门。放线菌门和子囊菌门(Actinobacteria)分别为不同处理组的优势菌门(图3e,图3f)。

图3 不同处理组丰富和稀有群落的优势门分布Fig.3 Dominant phylum distribution of rich and rare communities in different treatment groups
2.2.2 不同植被类型土壤丰富和稀有细菌、真菌群落多样性
土壤细菌、真菌α 多样性(Shannon)计算结果如图4 所示,总体来看,不同植被恢复类型显著改变了丰富和稀有微生物群落Shannon 指数的变化。丰富、稀有细菌和稀有真菌类群所有处理中,CK 组Shannon 指数显著低于植被恢复组(P< 0.05),而不同植被恢复类型对丰富真菌群落的Shannon 指数无显著影响。在稀有细菌群落中GL 的Shannon 指数显著高于CF(P< 0.05),而BL、BF、和MF 组无显著差异(图4b)。GL 和BL 组中稀有真菌的Shannon 指数较大(图4d),显著大于CF、BF 和MF 组(P< 0.05)。然后,对丰富和稀有细菌、真菌在ASVs 水平上进行基于Bray–Curtis 距离的PCoA 分析,结果如图5 所示,不同处理组的丰富和稀有细菌、真菌Beta 多样性表现为组间分离、组内紧密,形成明显的聚类效应,表明不同植被恢复处理及CK 之间群落Beta 多样性差异显著。丰富、稀有细菌中,CK 与其他植被恢复组沿X轴呈分离趋势,表明CK 和其他植被处理组的群落结构间存在显著差异(P< 0.05,图5a 和5b),而丰富细菌中不同植被恢复组间相互重叠,差异不明显,说明这些处理间丰富细菌群落系统发育更为接近。CK、GL 和BL 组丰富和稀有真菌群落沿x轴与CF、BF 和MF 组之间存在距离(图5c 和5d)。CF、BF 和MF 组稀有真菌群落趋势圈相近,几乎重叠,但CK 沿y轴与GL 和BL 组有一定距离。

图4 不同处理组对α 多样性的影响Fig.4 Effects of different treatment groups on α diversity
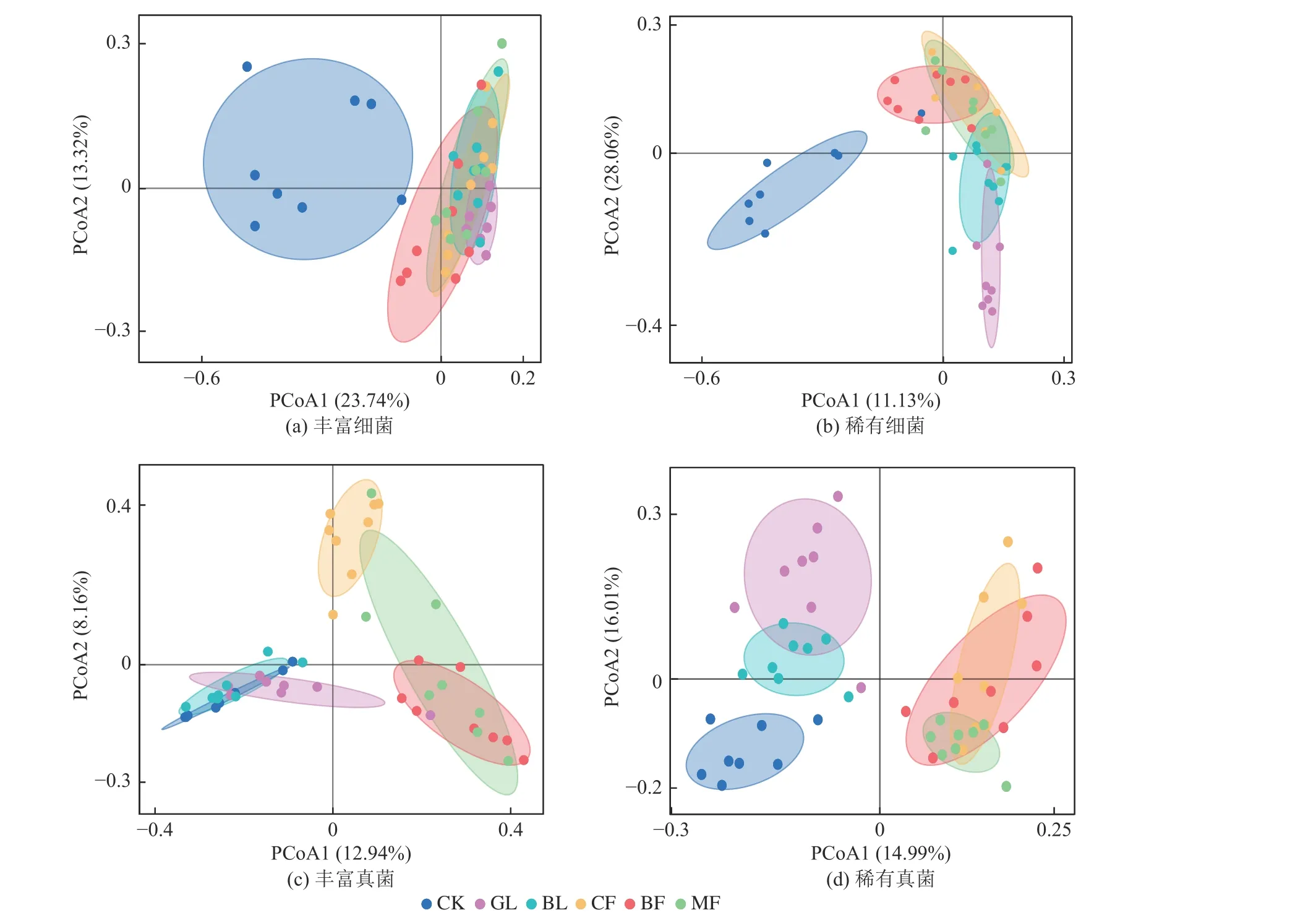
图5 不同处理组土壤细菌、真菌β 多样性Fig.5 β diversity of soil bacteria and fungi in different treatment groups
2.3 不同植被类型土壤丰富和稀有类群共现网络
基于ASVs 之间的Spearman 相关性,构建丰富与稀有微生物共现网络,探索不同植被恢复模式下丰富、稀有微生物群落内部物种间的互联关系。基于随机矩阵理论的方法,相关系数的阈值确定为0.65,分析所构建每个网络的拓扑属性(表1 和表2)。表1 显示,与CK 处理相比,植被恢复增加了丰富和稀有细菌网络的节点数(nodes)、连接数(links)和网络度(degree)等拓扑属性。GL、BL、CF、BF 和MF丰富细菌网络分别由140、167、134、138、139 个ASV 节点和471、633、545、601、588 条连接构成,高于CK(112 个节点和442 条连接)。GL、BL、CF、BF 和MF 稀有细菌网络分别由279、317、258、267、273 个ASV 节点和1 771、1878、1 471、1 783、1 973条连接构成,远高于CK(172 个节点和893 条连接)。BL 丰富真菌网络由92 个ASV 节点和183 条连接构成,远高于CK(64 个节点和124 条边),而其余恢复类型下丰富真菌网络节点和连接数均略高于CK网络(表2)。BL、BF 和MF 稀有真菌网络分别由280、218、240 个节点和1 490、932、1 008 条连接构成,远高于CK 稀有真菌网络(102 个节点和243 条边)。总体上,不同植被恢复类型均提高了丰富和稀有细菌、真菌的网络拓扑参数和复杂度。

表1 不同植被恢复模式土壤细菌共现网络的拓扑属性Table 1 Topological properties of soil bacteria co-occurrence networks in different vegetation restoration modes
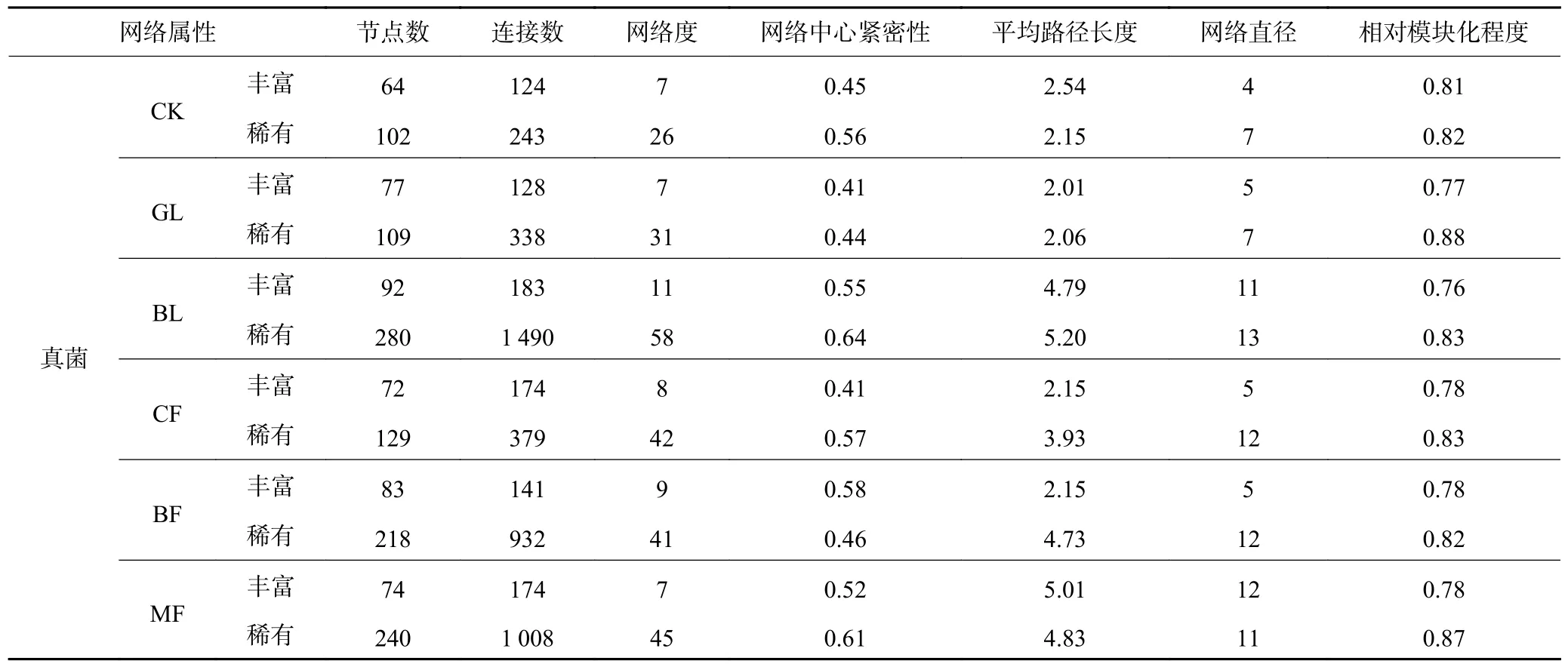
表2 不同植被恢复模式土壤真菌共现网络的拓扑属性Table 2 Topological properties of soil fungi co-occurrence networks in different vegetation restoration modes
图6 的共现网络分析结果表明:不同植被恢复类型的网络结构比CK 更加复杂,尤其是GL、BL、CF、BF 和MF 稀有细菌网络,和BL、BF 和MF 稀有真菌网络。丰富细菌中Actinobacteria、Proteobacteria、Acidobacteria和Chloroflexi4 个菌门占共现网络ASV 节点>90%,其中:Actinobacteria(44.27%~46.44%)和Proteobacteria(24.54%~28.36%)占据绝对主导地位。除了BF,其他植被恢复网络节点之间正向关系占比大于70%,且高于CK,菌群之间更趋向于合作。图6b 中,稀有细菌中Armatimonadota、Cyanobacteria和甲基米拉氏菌门(Methylomirabilota)3 个菌门占共现网络ASV 节点>76%,处于主导地位。虽然所有植被恢复网络节点之间正向关系占比大于50%,菌群之间趋向于合作关系,但正向关系占比均小于CK。丰富真菌网络中,Ascomycota和Basidiomycota在共现网络ASV 节点中占据主导地位,占比>77%,且所有网络连接以正向关系为主,其占比大于85%,菌群之间趋向于合作关系。稀有真菌网络中,Rozellomycota和梳霉门(Kickxellomycota)在所有网络ASV 节点中占比>69%,除了MF 的占比为52%。所有稀有真菌网络连接以正向为主,均大于88%。

图6 不同植被类型细菌与真菌微生物共现网络图Fig.6 Co-occurrence network of bacteria and fungi in different vegetation types
为进一步确认微生物网络对干扰的抵抗力(稳健性)强弱,通过移除节点改变自然连通性的幅度进行测试,结果如图7 所示,去除相同比例的节点后,所有植被恢复模式下的丰富和稀有细菌、真菌网络的自然连通性比原始状态均有所下降,而综合来看,CK 斜率最大,下降速率最大,说明其微生物网络受环境干扰抵抗力较弱。对于丰富细菌和丰富真菌网络(图7a 和图7c),BL 变化幅度较小,GL 波动次之,表明BL 和GL 增加了土壤丰富细菌和真菌网络的稳定性。稀有细菌和稀有真菌网络(图7b 和图7d)中,GL 和BL 的变化幅度最小,说明GL 和BL 分别提高了土壤稀有细菌和稀有真菌网络对外界干扰的抵抗能力。

图7 不同植被类型微生物自然连通性与移除节点比例之间的关系Fig.7 Relationship between natural connectivity of microorganisms and the proportion of removed nodes in different vegetation types
2.4 不同植被类型环境因子对土壤丰富和稀有微生物群落相关性分析
将土壤微生物中丰富细菌、稀有细菌、丰富真菌和稀有真菌群落结构指标分别作为一个矩阵,依次与土壤理化性质与酶活性指标做Mantel 测试,结果表明不同植被恢复类型下丰富和稀有类群均不同程度受到土壤理化和酶活性的显著影响(P< 0.05),而CK 组仅有丰富细菌和稀有细菌受到ALP 和BG 酶活性影响(图8)。图8b 显示,GL 恢复组中URE 酶极显著影响丰富细菌群落组成(P< 0.01)。BL 处理组中pH、SOM、AP、BG 和ALP 均与稀有细菌群落呈显著性差异(P< 0.05),pH 和AP 与丰富细菌群落有显著关系(P< 0.05),而NN 极显著作用于丰富细菌群落(P< 0.01),而URE 则与丰富和稀有真菌群落均具有显著相关性(P< 0.05)(图8c)。图8d 中,CF恢复组的稀有真菌群落与LAP 酶活性呈极显著相关,而BF 恢复组的URE 酶活性则与土壤细菌、真菌丰富和稀有菌群落组成均具有极显著相关性(P< 0.01),SOM 显著作用于丰富细菌群落(图8e)。图8f 显示,URE 和ALP 酶活性极显著作用于MF 恢复组的土壤丰富和稀有细菌群落(P< 0.01)。
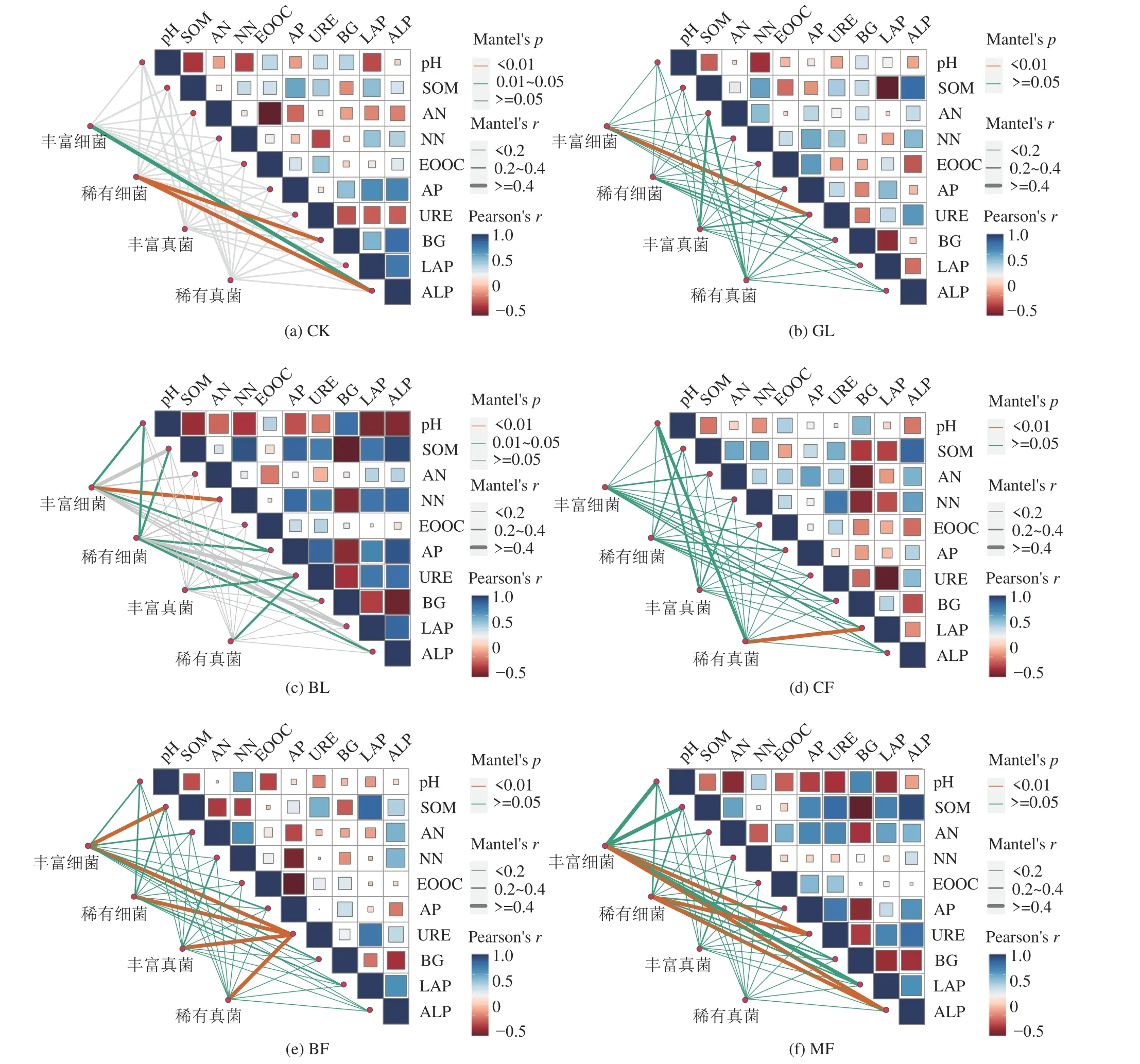
图8 不同植被类型土壤理化性质、酶活性与丰富、稀有细菌与真菌分类群群落结构相关性Fig.8 Soil physical and chemical properties, enzyme activities and community diversity of abundant, rare bacteria and fungi were correlated with different vegetation types
3 讨 论
3.1 矿区不同植被恢复类型对土壤理化性质和酶活性的影响
研究表明,不同植被恢复类型对土壤和酶活性的影响差异较大(图2)。不同植被的根系分泌物和生理代谢存在明显差异,因此植被类型的变化通常被认为是改变土壤理化性质和酶活性的重要因素[19]。BL 恢复组中有机质、硝态氮和有效磷含量高于其他处理组,而pH 显著低于其他处理组(图2),这可能归结于土壤有机质主要来源于地表植被残体、凋落物、地下坏死的根系及其分泌物和微生物残体等,沙棘属于落叶性灌木,根系较为发达,有利于增加土壤中有机质含量。此外,沙棘是干旱地区少数具有固氮能力的木本植物,虽然不是豆科植物,但它可与某些固氮菌共生,这些固氮菌在沙棘的根系上生长,形成根瘤,来固定大气中的氮[20],这可能是BL恢复组土壤中硝态氮含量较高的原因。有研究报道[21],沙棘根系分泌物中酸类物质含量最高,导致土壤pH 值较其他处理组偏低,而土壤pH 能够影响土壤磷元素的化学形态,在低pH 值的土壤中,无机磷组分变化较大,使得有效磷含量增加。胞外酶是由微生物分泌到土壤中,可将复杂有机化合物降解为可被吸收的小分子,并从土壤中吸收生存所需的营养和能量,对微生物分解有机质及植物对土壤碳、氮、磷等营养元素的吸收利用发挥调节作用,在土壤生态系统物质循环和能量交换中起着重要作用[22]。研究发现不同植被恢复类型对土壤胞外酶活性影响存在显著性差异,所有植被处理组的β-葡萄糖苷酶活性均显著低于CK,说明植被恢复组的可用碳源已满足微生物生长需要,因为微生物只有在可用营养物质稀缺时才会以生长和代谢为代价生产酶。BL 的脲酶和碱性磷酸酶活性显著高于其他植被恢复组,脲酶(URE)等与土壤氮矿化和循环密切相关,能够表征土壤中氮素营养状况,磷酸酶(ALP)主要参与磷的代谢和转化,BL 土壤中硝态氮和有效磷含量高于其他处理组,促使参与氮磷循环相关微生物活动增强,这使得URE 和ALP 酶活性增加,应对氮磷营养的消耗,在土壤环境变化时维持养分平衡。宁岳伟等[23]在山西大同矸石山不同植被复垦区土壤酶活性研究中发现,云杉林土壤具有最高的脲酶和碱性磷酸酶活性、以及较好的氮素营养和有效磷含量,这与研究结果高度一致。
3.2 矿区不同植被类型对土壤丰富和稀有微生物群落多样性和组成的影响
土壤微生物群落与地上植被类型具有密切相关性,不同植被由于凋落物生物量和分解速率不同,以及根系分泌物和根系脱落物性质不同,可以直接影响土壤微生物群落结构分布,还可通过改变土壤理化性质间接影响土壤微生物,因此,不同植被会通过直接和间接作用,选择性地吸引一些特定微生物,使得这类微生物在林下土壤富集,从而导致微生物群落结构发生变化[24]。此外,一些研究表明,丰富和稀有微生物类群在多样性与群落组成方面对环境因子变化的响应亦不一致,故在不同的植被类型中可能会表现出偏向的丰度分布。研究发现放线菌门(Actinobacteria)、变形菌门(Proteobacteria)、绿弯菌门(Chloroflexi)和酸杆菌门(Acidobacteria)是该矿区不同植被恢复类型土壤中优势丰富细菌门类(图3a),可能是由于这些门类具有较好的环境适应性,且具有较高生态位的缘故。放线菌门是土壤中纤维素和木质素的主要分解者,在贫瘠的土壤中亦能较好生存[25]。变形菌门是典型的碱性环境优势菌,且其具有固氮作用,可适应氮含量较为匮乏的环境,绿弯菌门和酸杆菌门亦被报道具有较强的生态适应能力[26-28]。BASTIDA 等[29]报道在半干旱矿区复垦土壤中放线菌门最为丰富,且KONG 等[30]和LI 等[31]也在干旱和半干旱矿区复垦土壤中发现变形菌门和放线菌门是优势细菌类群,与研究结果一致,这说明矿区复垦土壤中丰富细菌具有较好的环境适应能力,并未对植物类型产生较大的依赖性。JIAO 等[32]认为,丰富物种通常在生态系统中占据更为广泛的生态位宽度,可以利用更多的资源,这使它们比稀有物种更能适应外界环境扰动。稀有细菌分类群中,装甲菌门(Armatimonadota)和蓝藻门(Cyanobacteria)为优势稀有细菌门(图3b)。Armatimnadota属于好氧型寡营养菌门,可以降解一些复杂的碳化合物,其新成员被报道有可能生成乙酰乙酸,参与氮和硫循环,并介导海洋沉积物中碳循环关键过程[33]。王泽铭等[34]报道Armatimonadota为干旱区草地土壤的优势菌属之一,李文宝等[35]亦是发现Armatimonadota是寒旱区典型草地土壤细菌群落结构组成中的关键菌属,与本文结果类似。GL 处理组的脱硫杆菌门(Desulfobacterota)丰度显著高于其他植被复垦组,推测可能是草本植被苜蓿特殊的根系分泌物造成的根际环境使得脱硫杆菌门丰度偏高,脱硫杆菌门可将有机质完全氧化成CO2或部分氧化为乙酸盐,降低碳源利用能力,抑制有机氮矿化,GL 恢复组的有机质和硝态氮含量偏低可能与此相关[36]。子囊菌门和担子菌门是自然界真菌群落中分布最广和丰度最高的 2 个真菌类群,多数为腐生菌,在多种生境中具有较强的生存能力。本研究中子囊菌门和担子菌门为植被复垦土壤丰富真菌的主导菌门(图3c),与YANG 等[37]在黄土高原半干旱区退耕草地中研究的结果相似。研究中样地土壤pH 为7.7 左右,由于碱性土壤适宜腐生真菌生长,这可能是子囊菌门和担子菌门为优势丰富菌门的一个原因[27]。研究中BL、CF、BF 和MF 恢复组的稀有真菌门罗兹菌门(Rozellomycota)相对丰度占比最高,BAI 等[28]发现板栗和茶树间作模式下,土壤中Rozellomycota丰度较高与研究结果类似。此外,植被恢复对于提高丰富和稀有微生物多样性具有重要作用。较CK 处理组,植被恢复组丰富和稀有细菌及稀有真菌类群多样性较高,在矿区排土场复垦过程中,BL 在干旱半干旱地区为先锋物种,植被的生长可能为土壤微生物活动提供大量营养物质和良好的生存环境,这可能解释了BL 中微生物的多样性略优于其他植被复垦组。且稀有细菌和真菌类群的多样性高于丰富类群,可能是因为稀有微生物类群是微生物重要的种子库,可提供巨大的遗传多样性和功能多样性储备,是土壤微生物多样性的主要贡献者,这恰与以往的研究结果相吻合[38-39]。
3.3 矿区不同植被类型土壤丰富和稀有微生物群落网络特性及稳定性分析
微生物不是孤立地生活在土壤中,相互之间具有较为复杂的关系,且物种间共生或竞争模式在一定程度上可以影响土壤微生物群落的结构和功能[29]。共现网络结果中(表1 和表2,图6),相较于对照组,植被恢复组增加了丰富和稀有细菌与真菌网络的节点数、总连接数、和整体网络度等拓扑属性,说明植被复垦土壤中微生物网络规模更为复杂,节点间的联系更为紧密,使得群落间物质、能量和信息的传递效率高,助力微生物群落更加有效地利用土壤养分。BL 在所有处理组中表现出较高的微生物网络复杂性,这是因为沙棘根蘖能力强,可以固氮,加上根系分泌的有及酸类物质可以改善根际土壤状况,随着根系的扩展,微环境分散,为微生物生长提供了更多的生态位,也可能为其他微生物提供了更多生存机会,引发更为丰富和复杂的生态相互作用,而这种复杂性通常涉及更多微生物群落之间的物质循环、能量流动和信息传递等方面,这一结果与YUAN 等报道土壤微生物多样性增加改善网络复杂性的研究相一致[40]。此外,结果显示稀有类群比丰富类群具有更高的网络度和中心紧密性(表1 和表2,图6),这表明稀有细菌、真菌分类群间存在更为复杂的耦合关系,网络复杂性的增加会提升网络稳定性和应对外界干扰的抵抗性,这对于维持生态系统稳定至关重要[41-42]。有理论研究认为,网络鲁棒性可以通过“攻击”边缘或节点后的自然连接来衡量,攻击下自然连接的抵抗力更大,表明网络更稳定[43]。基于自然连通性的抗损伤性测试也表明(图7),BL 提高了丰富细菌、丰富和稀有真菌网络节点抵抗干扰的能力,说明沙棘恢复模式土壤微生物群落具有更高的稳定性,而网络拓扑参数结果中BL 网络具有较长的平均路径同样说明其响应环境变化的速度较慢,群落稳定性较高,这可能是BL 中土壤养分含量较高,有利于微生物,尤其是真菌的新陈代谢和繁殖[32,41],提高各类群在资源有限的矿区土壤环境中的种间竞争和协作关系,复杂的相互作用有助于形成更为健康的微生物区系,进而提升微生物群落网络的交互度和稳定性,更好的保障了微生物生态功能发挥。
3.4 土壤微生物群落与环境因子的互作机制
研究结果表明,不同植被恢复类型下丰富和稀有类群受到不同土壤理化和酶活性的显著影响(图8,P< 0.05)。多种土壤环境因子(pH、SOM、AP、URE等)显著影响植被恢复土壤中丰富群落分布,与Pan等[44]在草地土壤中得到的结果一致。丰富类群比稀有类群对环境因子敏感性更低,可能是因为丰富类群在环境因子主导下的群落演替是定向收敛的,进而占据土壤微生物群落的主导地位和更宽的生态位,使得它们在资源竞争方面更具优势,从而对矿区环境有较高的适应性,在环境扰动中保证了优势地位,刘蕾等[5]报道丰富细菌是养分、能量代谢等基础功能的主体参与者,这与研究结果恰好能对应上。文中稀有群落亦是与pH、SOM、AP、BG 和ALP 等环境因子具有显著相关性,这可能是因为稀有群落具有较高的多样性,使其在面临环境变化时做出快速响应以适应新环境,以拓宽其狭窄的生态位和提升其资源竞争能力。
4 结 论
1)矿区排土场植被复垦显著地改善了土壤理化性质和酶活性。其中BL 对土壤SOM、NN 和AP 积累有优势,并且显著提高了土壤URE、LAP 和ALP酶活性。
2)不同植被恢复模式对土壤丰富和稀有细菌、真菌群落结构组成有显著影响。丰富细菌中放线菌门最为丰富,且在CK 中占比最高,显著高于其他植被恢复组(P< 0.05),装甲菌门和蓝藻门为共同优势稀有细菌门,子囊菌门和担子菌门为真菌群落主导菌门,BL、CF、BF 和MF 组的罗兹菌门相对丰度占比最高。不同植被恢复类型丰富和稀有细菌、真菌群落Shannon 指数高于CK。
3)不同植被恢复类型提高了丰富和稀有细菌、真菌的网络拓扑参数,如节点数、连接数和网络度等。共现网络分析显示,不同植被恢复类型的网络结构比CK 更加复杂,尤其是GL、BL、CF、BF 和MF 稀有细菌网络,和BL、BF 和MF 稀有真菌网络。自然连通性的幅度测试结果表明,所有植被恢复模式下的丰富和稀有细菌、真菌网络稳定性和受环境干扰抵抗力优于CK,尤其BL 对土壤丰富细菌、丰富真菌和稀有真菌网络的稳定性提升度最高。
4)不同植被恢复类型下丰富和稀有类群均不同程度受到土壤理化和酶活性的显著影响(P< 0.05)。CK 组仅有丰富细菌和稀有细菌受到ALP 和BG 酶活性影响。BL 恢复组中pH、SOM、AP、BG、URE、ALP 与富有和稀有细菌和真菌群落具有显著相关性。
猜你喜欢
昆明医科大学学报(2022年2期)2022-03-29 00:51:58
食品安全导刊(2021年20期)2021-08-30 06:40:50
中国比较医学杂志(2020年4期)2020-05-26 05:47:22
江苏农业科学(2019年5期)2019-09-02 14:01:46
水生生物学报(2019年4期)2019-07-20 08:08:10
生物安全学报(2019年3期)2019-02-15 16:54:12
川北医学院学报(2019年6期)2019-02-10 10:48:32
广东农业科学(2017年5期)2017-08-29 10:37:31
水生生物学报(2015年1期)2015-02-28 16:01:05
应用海洋学学报(2014年1期)2014-11-22 07:17:44
