
获得性免疫在干眼的启动与维持
2024-04-07 01:16孙文鑫郭俞利刘祖国
国际眼科杂志 2024年4期
孙文鑫,郭俞利,刘祖国,3
0引言
干眼(dry eye,DE)是多因素引起的慢性眼表疾病,是由泪液的质、量及动力学异常导致的泪膜不稳定或眼表微环境失衡,可伴有眼表炎症反应,组织损伤及神经异常,造成眼部多种不适症状,严重时可引起视功能障碍[1]。目前,我国按照泪液的主要成分和泪液动力学因素将干眼分为5类,分别是水液缺乏型干眼、脂质异常型干眼、黏蛋白异常型干眼、泪液动力学异常型干眼及混合型干眼[2]。既往研究表明,DE是一种炎症性疾病,与自身免疫性疾病有许多共同特征。相关基础研究表明,干眼是眼表的非感染性炎症[3]。环境因素、内源性应激和抗原等应激反应被认为是DE的触发机制。眼表免疫系统被激活,免疫细胞浸润到眼表组织,分泌多种免疫分子造成眼表炎症反应,加剧眼表损伤。本文就干眼的免疫机制研究进展进行综述并重点讨论获得性免疫在DE的启动与维持。
1天然免疫
角膜上皮细胞位于眼表的最外层,并与环境直接接触[4]。长期暴露于多种环境应激下引起泪膜不稳定性增加最终导致眼表高渗状态,引起角膜上皮细胞损伤和天然免疫炎症反应发生,最终导致眼表疾病[5]。环GMP-AMP合成酶(cGAS)刺激因子(STING)通路是近年来通过识别细胞质DNA发现的一种炎症信号通路[6-7]。在各种应激条件下,来自细胞核和线粒体的DNA释放到细胞质中[8],cGAS识别细胞质双链DNA(dsDNA)并产生称为2’3’-cGAMP的第二信使,该信使进一步激活STING的表达和易位,诱导TBK1(p-TBK1)和IRF3(p-IRF3)磷酸化,进一步介导下游炎症细胞因子(IFN-a/β和CXCL10)的释放[9]。我们团队的最新研究发现,高渗应激(HS)介导线粒体DNA释放到细胞质中可能通过cGAS-STING信号通路引起炎症。这项研究提供了线粒体DNA感应激活cGAS-STING信号通路的证据,该通路在两个实验性干眼模型(BAC诱导的小鼠眼表损伤模型和手术摘除泪腺的干眼小鼠模型),干眼患者样本以及暴露于HS培养的HCE中介导眼表炎症反应[10]。这一研究证实了角膜上皮细胞在DE炎症反应启动中的新作用,这一与天然免疫反应相关的信号通路的识别,可能成为DE潜在的治疗靶点。此外,眼表的高渗状态早期通过激活丝裂原活化蛋白激酶(MAPK)和核因子κB(NF-κB)应激信号通路,启动一系列反应破坏角膜上皮细胞屏障功能。产生的活性氧(ROS)抑制了磷脂酰肌醇3激酶(PI3K)/蛋白激酶B(AKT)信号转导[11],导致眼表上皮细胞受损或死亡,同时通过Toll样受体4(TLR4)、Nod样受体蛋白-3(NLRP3)炎症小体等向天然免疫细胞传递炎症信号,产生炎症因子导致眼表炎症[12]。大量研究表明,干眼状态下泪液和角膜组织中炎症细胞因子如IL-1β、TNF-α和IL-6等表达水平增高[13]。
干眼导致眼表组织发生损伤后,角膜上皮细胞首先启动了免疫炎症反应,随后眼表的中性粒细胞、巨噬细胞、自然杀伤细胞等先后活化,这些天然免疫细胞的活化,一方面扩大了眼表炎症级联反应,造成眼表损伤;另一方面促进了自身反应性T细胞的活化与分化,激活获得性免疫反应。T细胞活化后又可以分泌多种炎症因子和趋化因子继续促进天然免疫反应的发生,形成恶性循环(图1)。因此,了解DE发生发展时的病理过程有助于为DE的治疗提供新靶点。
2获得性免疫
获得性免疫(acquired immunity)是指体内抗原特异性T/B淋巴细胞接受抗原刺激后,自身活化、增殖、分化为效应细胞,产生一系列生物学效应的全过程(图2)。根据参与免疫应答细胞种类及其机制的不同,可将获得性免疫反应分为B细胞介导的体液免疫应答和T细胞介导的细胞免疫应答两种类型[14]。T细胞是不均一的细胞群体,根据其表面标志及功能特点,可分为不同的T细胞类别及其亚群。在本篇综述中,我们根据T细胞抗原受体(TCR)分类将T细胞分为TCRαβT细胞和TCRγδT细胞,并分别介绍这两类T细胞介导的免疫反应在DE病理中的作用。
2.1TCRαβT细胞细胞免疫是人体免疫系统的重要组成部分,在清除癌变组织和病原体入侵中发挥重要作用。在既往临床和基础研究中观察到DE患者和小鼠结膜中有T细胞的浸润,提示T细胞参与了干眼的病理过程[15],但具体的T细胞亚群特征及功能有待阐明。近期一项研究特异性地分离了DE小鼠的结膜,并通过高通量TCR测序来揭示DE发生时的TCR序列。与对照组小鼠相比,干眼小鼠结膜αβT细胞受体(αβTCR)的α链和β链克隆型明显增加。TCRα链和β链的互补决定区3区(CDR3)氨基酸长度较健康对照组显著增多[16]。αβT细胞亚群占T细胞群体的65%-70%,其表面的TCRs由α链和β链两条糖蛋白链组成,具有主要组织相容性复合物(MHC)介导的特异性识别抗原细胞毒功能[17]。该研究进一步证实了DE是一种主要由T细胞介导的自身免疫性疾病,αβT细胞亚群参与了DE的发展并为DE的治疗提供了新靶点。
目前关于DE获得性免疫应答的研究主要集中在CD4+T细胞。T细胞在眼表浸润的场所主要为结膜。CD4+T细胞可以通过淋巴细胞功能相关抗原1(LFA-1)黏附在眼表组织中并促进DE炎症发生现已成为DE的治疗靶点[18]。一项来自新加坡的临床研究对健康个体和DE患者结膜样本中的T细胞进行了分析,研究者通过收集结膜印记细胞学样本对来自健康供体的T细胞进行分析发现CD8+T细胞对CD4+T细胞具有明显的优势[19]。此外,研究还发现健康人的结膜中含CD8+/CD69+/CD103+/CCR7-T淋巴细胞。利用一系列T细胞特异性的表面标记物,研究者进一步分析了52例DE患者眼表存在的不同T细胞亚群。根据特异性标记物,可以将这些DE患者分为两种类型:第一类DE患者有较高的眼红发生率,结膜中CD8+T细胞的数量明显升高;第二类DE患者的泪膜不稳定性增加,结膜中CD4+T细胞的比例更高,研究结果表明大部分患者属于第二类。因此,与健康眼部组织样本中的T细胞相比,DE发生时T细胞群的再分配导致了其发病机制的复杂性[19]。近期的一项基础研究评估了CD4+T细胞在苯扎氯铵(BAC)诱导的C57BL/6小鼠眼表损伤中的致病作用。研究结果表明,与正常组相比,通过BAC诱导可以引起CD4+CD69+,CD4+IFN-γ+和CD4+IL-17+细胞数量增加,并且伴有IFN-γ、IL-17、Th1、Th17以及转录因子T-bet和RORγt的增加。此外,BAC还可以引起眼表损伤,包括角膜屏障功能受损,结膜杯状细胞丢失和泪液生成减少。从BAC诱导的小鼠体内特异性地分离出CD4+T细胞并过继转接到裸鼠体内,可导致与直接局部BAC处理相似的眼表表现,具体包括了CD4+T细胞、IFN-γ、IL-17的增加以及眼表损伤[20]。干燥综合征相关DE(ssDE)发生时伴有泪腺、结膜和睑板腺上皮细胞的自身免疫性炎症反应,并且产生的CD8+T细胞可以通过穿孔素-颗粒酶途径破坏角膜上皮[21]。由此可见,深入研究CD4+T细胞和CD8+T细胞在DE发生发展中的具体作用,对于DE免疫炎症方面的治疗具有重要指导意义。
辅助性T细胞17(T helper cell 17,Th17)是一种能够分泌白介素17(interleukin17,IL-17)的T细胞亚群,在自身免疫性疾病和机体防御反应中具有重要的意义。Th17细胞在淋巴组织中分化扩增后迁移到眼表并释放炎症细胞因子发挥致病作用,引起角膜和结膜上皮细胞凋亡,睑板腺基底腺泡细胞增殖和脂质生成异常,以及神经退行性改变[22]。随着急性期的消退,记忆性Th17细胞持续产生的IL-17可维持DE发生时眼表慢性炎症反应,局部阻断IL-17可显著缓解DE症状[23]。Th17细胞除了可以直接引起眼表组织损伤外,还可以通过放大炎症反应介导DE病理过程导致恶性循环,IL-17可以有效地诱导内皮细胞和上皮细胞分泌IL-6、TNF-α、IL-1β和IL-8,引起眼表炎症级联反应,进一步招募中性粒细胞和巨噬细胞并激活抗原提呈细胞(antigenpresenting cells,APCs)[24]。在由过敏性疾病导致的睑板腺功能障碍的小鼠模型中发现Th17细胞介导的中性粒细胞向结膜聚集,在导致睑板腺阻塞中起核心作用,阻断Th17细胞免疫反应可以显著减少睑板腺阻塞的发生[25]。同时APCs和CD4+T细胞通过免疫突触在淋巴结中相互作用,产生大量促炎细胞因子,刺激CD4+T细胞分化为不同类型的T细胞亚群。Th17产生的粒细胞-巨噬细胞集落刺激因子(GM-CSF)主要通过促进眼表APCs的成熟和迁移促进DE眼表炎症反应,有研究表明DE发生时泪液中GM-CSF水平升高[26]。此外,免疫系统也会随着年龄的增长而发生着各种变化,导致全身各个系统慢性低水平炎症状态和自身免疫的风险增加[27]。由于DE在中老年患者中更为普遍和严重。因此可以推测Th17细胞免疫反应的发生和衰老之间具有密切的联系。同时有研究发现随着年龄的增长,Th17细胞免疫反应增强[28]。Th17细胞一方面从淋巴组织迁移到眼表发挥致病作用,另一方面通过参与角膜淋巴管生成促进APCs从眼表运输到引流淋巴组织激活免疫反应,同时还可以促进生发中心的形成,诱导B细胞增殖,因此具有高度致病性。
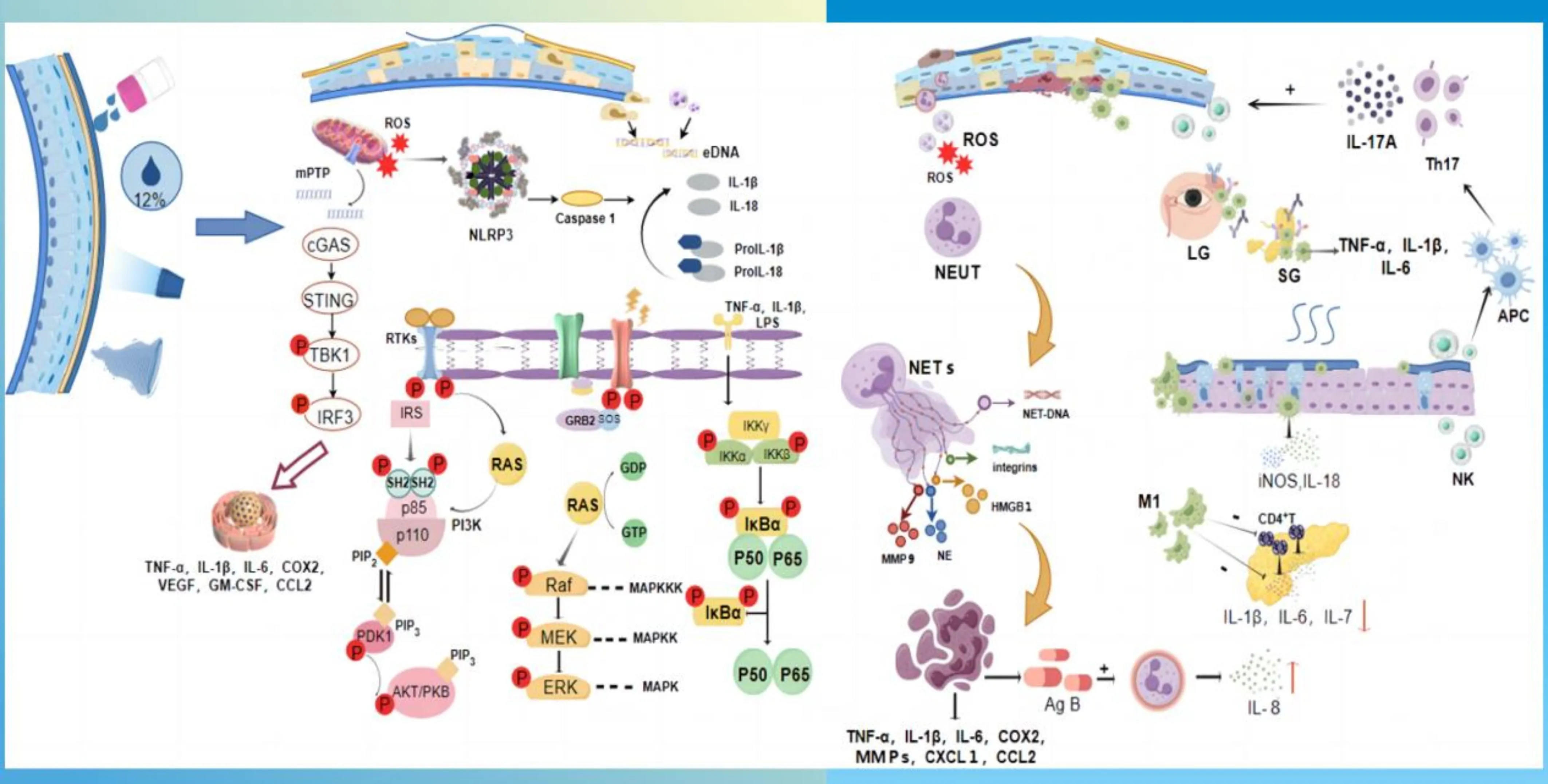
图1 天然免疫在DE病理发展中的作用(本图由Figdraw绘制)。
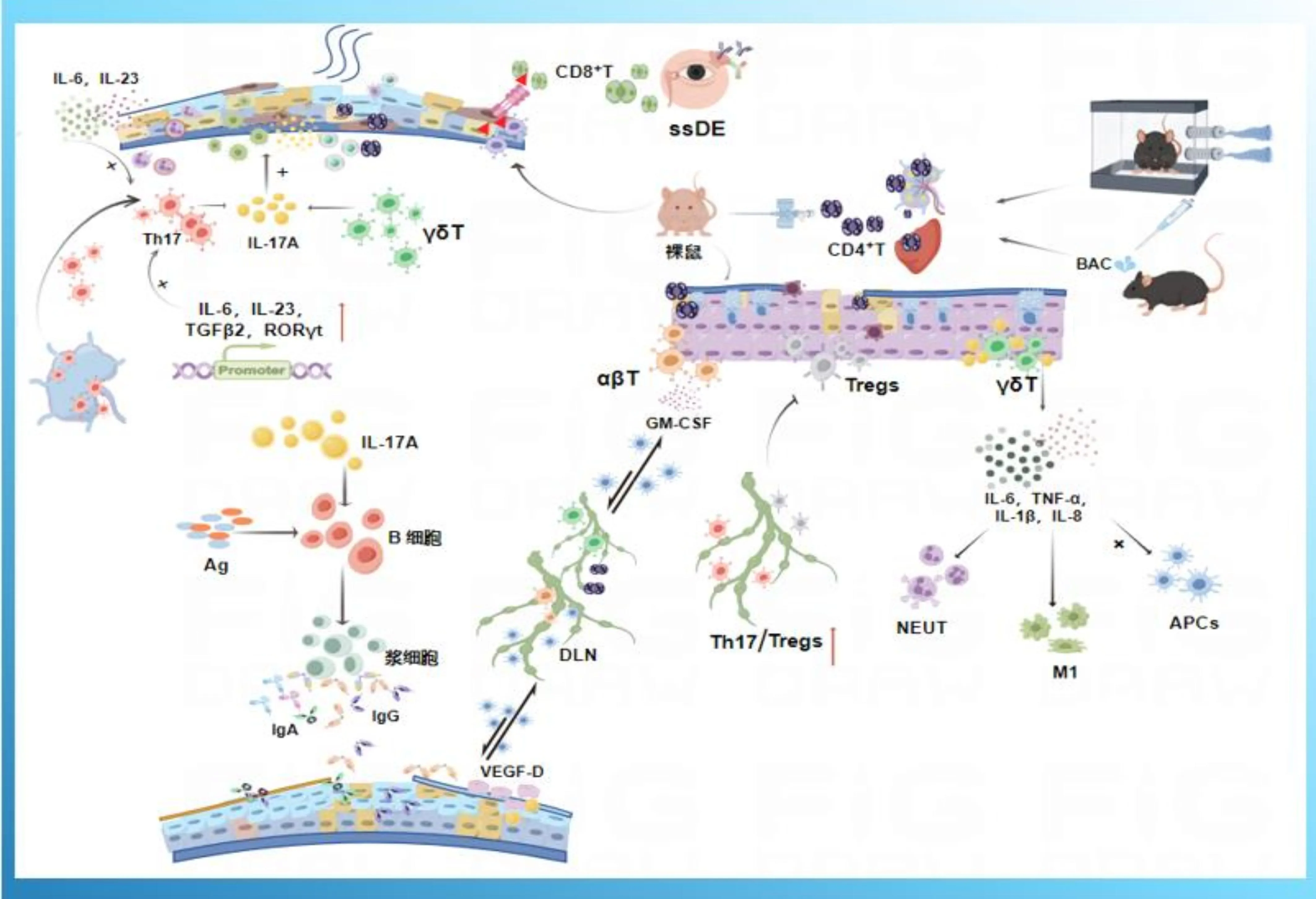
图2 获得性免疫在DE病理发展中的作用(本图由Figdraw绘制)。
调节性T细胞(regulatory T cell,Treg细胞)作为眼表微环境的重要组成部分,可以通过多种机制抑制免疫炎症反应的形成。Treg细胞在诱导机体免疫耐受,调节机体免疫平衡方面起着重要作用。Treg细胞免疫防御功能受损或数量减少会破坏眼表免疫稳态进而引起多种眼表疾病。近年来,越来越多的研究证据表明,Treg细胞相关免疫疗法在DE治疗中具有潜力。有研究发现,增加泪腺中趋化因子CCL22的局部释放来诱导内源性Treg细胞聚集可以有效减少淋巴组织中CD4+T细胞的浸润,减少角膜上皮荧光素钠染色,增加泪液分泌和结膜杯状细胞数量。局部增加功能正常的Treg细胞数量改善免疫失衡可以有效减轻实验性动物干眼模型中免疫炎症反应的发生,从而缓解DE相关眼表症状[29]。此外,静脉注射色素上皮衍生因子(PEDF)和间充质干细胞及其外泌体疗法可以抑制Th17细胞并促进Treg细胞增殖,增强免疫抑制功能,减轻DE严重程度[30]。由此可见,Treg细胞参与了获得性免疫反应,并在DE病理进展中发挥重要作用。
2.2TCRγδT细胞γδT细胞是T细胞的一个亚群,占外周血液T细胞总数的5%以下,主要分布在皮肤、小肠、食管、肺、生殖器及皮下组织,是皮肤表皮内淋巴细胞和黏膜组织上皮内淋巴细胞的主要成分之一[31-33]。γδT细胞虽然具有T细胞表面受体,但在功能上主要参与天然免疫反应,可以不受MHC限制而识别多肽抗原和磷酸盐抗原[34]。研究表明,γδT细胞是病毒感染早期受累器官中的重要细胞因素,在中枢神经系统(脑膜炎)和肠道(结膜炎)等自身免疫性疾病的炎症中扮演重要角色[35]。γδT细胞作为重要的效应性T细胞,目前对其生物学意义的认识尚处于起始阶段。既往研究表明:Th17细胞在IL-17A介导的干眼免疫发病机制中发挥主要作用。陈蔚教授团队近期研究发现,正常小鼠结膜中分泌IL-17A的细胞分群中,高达近60%的细胞是γδT细胞;同时结膜中γδT细胞比例与干眼的严重程度呈正相关;消除γδT细胞后,干眼小鼠眼表症状减轻[36]。该研究评估了人类和实验性小鼠DE中γδT细胞介导的炎症反应,并强调了靶向IL-17和γδT细胞在DE治疗中的潜在协同作用。
2.3B淋巴细胞B淋巴细胞简称B细胞,是由骨髓中的造血干细胞分化发育而来。B细胞在抗原刺激下可分化为浆细胞,在免疫监控和应对病原体入侵中扮演着核心角色[37]。为了维持眼表稳态,位于眼表的浆细胞可以不断地释放分泌性IgA,成为黏膜保护中最重要的体液成分之一。同时IgA还可以限制眼部微生物群侵入到深层组织。B细胞已被用于研究自身免疫性来源的眼部疾病,包括干燥综合征[38]和葡萄膜炎[39]。使用抗CD20抗体耗竭B细胞已被用于眼部疾病的治疗,这表明B细胞参与了自身免疫性疾病的发生与发展,但在DE免疫调节机制中的具体作用目前还没有明确的共识。
3小结与展望
眼部组织中包含了大量的免疫细胞,分布在眼表的免疫细胞维持了微环境的稳态,从而避免了不必要的组织损伤。近期有研究探讨了免疫炎症反应在DE中的作用,并重点讨论了DE、炎症和睑板腺功能障碍之间的恶性循环关系[40]。另外有针对DE发生的早期阶段进行的研究,发现神经肽在DE炎症中发挥着关键作用,通过抑制神经肽可能防止DE的恶化和进展[41]。上述研究对于DE的发病机制提出了新见解,但均没有对DE发生发展的全过程进行详细的描述。在本篇综述,我们描述了DE发生时眼表免疫反应的激活过程:当眼表的微环境稳态被破坏时,按照发生免疫炎症反应的时间顺序,角膜上皮细胞首先启动天然免疫反应,随后激活中性粒细胞、巨噬细胞等免疫细胞参与眼表损伤。当刺激因素持续存在或再次发生时,便开始启动获得性免疫引起T细胞和B细胞的聚集以及大量炎症因子的产生最终导致DE。在DE诊疗中,区分DE所处的免疫反应阶段对于DE的用药方案具有指导意义。对于轻度DE患者,主要采用物理治疗改善环境条件以减少天然免疫反应的发生,如减少屏幕使用时间、停止使用高气流风扇以及滴加人工泪液等。中度至重度DE患者则需要进行抗炎和免疫抑制等多靶点联合治疗减轻获得性免疫反应引起的眼表损伤。此外,免疫反应在DE病理进展的分子基础、标记物发掘、诊断试剂盒开发及相应阶段的治疗药物靶点研究是未来具有前景的研究方向。
猜你喜欢
基层中医药(2021年7期)2021-11-02
世界最新医学信息文摘(2021年12期)2021-06-09
中国生殖健康(2019年9期)2019-01-07
基层中医药(2018年10期)2018-12-06
基层中医药(2018年3期)2018-05-31
首都食品与医药(2018年3期)2018-03-18
科技视界(2018年33期)2018-02-21
西南国防医药(2016年6期)2016-12-01
保健与生活(2016年11期)2016-04-11
中国中医眼科杂志(2015年1期)2015-12-28
