
超高效液相色谱-串联质谱法快速测定蔬菜中4种异噻唑啉酮类化合物的含量
2024-04-02 07:22李根容田小艳余文琴
理化检验-化学分册 2024年3期
李根容,阮 燕,田小艳,余文琴,蔡 琼
(1.重庆市计量质量检测研究院,重庆 401123;2.国家农副加工产品及调味品质量监督检验中心,重庆 401123)
异噻唑啉酮类化合物是一类含有氮原子和硫原子的五元杂环化合物,主要包括5-氯-2-甲基-4-异噻唑啉-3-酮(CMIT)、2-甲基-4-异噻唑啉-3-酮(MIT)、1,2-苯并异噻唑啉-3-酮(BIT)、2-正辛基-4-异噻唑啉-3-酮(OIT)等。异噻唑啉酮类化合物是一种重要的灭菌防腐剂,因具有高效、作用持续时间长、可降解等优点被广泛应用于化工、农业园林、造纸、日化等领域[1-3]。研究表明,该类化合物具有细胞毒性和神经毒性,是一类致敏剂,可导致皮肤产生接触性过敏或皮炎等症状[4-5]。蔬菜中残留的异噻唑啉酮类化合物被人们直接摄入,危害身体健康[6]。近年来,国内外对玩具、化妆品、食品接触材料等日用品中异噻唑啉酮类杀菌防腐剂的使用制定了严格的法规标准[7-8],但我国现行国家标准尚未对蔬菜中的异噻唑啉酮类化合物的使用量和使用范围进行规定,仅在2015年农业部农药检定所起草发布的《农药助剂禁限用名单》(征求意见稿)中规定BIT、MIT的限量(质量分数)分别为0.1%、0.002 2%,但正式稿至今还未出台。因此,开发测定异噻唑啉酮类化合物含量的分析方法对保障蔬菜食品安全具有重要意义。
目前,异噻唑啉酮类化合物的检测方法主要有气相色谱法(GC)[9]、气相色谱-串联质谱法(GC-MS/MS)[10]、紫外分光光度法[11]、高效液相色谱法(HPLC)[12]、超高效液相色谱-串联质谱法(UHPLC-MS/MS)[13-16]等。HPLC灵敏度低,易出现假阳性,已经无法满足检验检测要求;GC和GC-MS/MS操作繁琐,分析时间长且容易造成目标物的损失。相较之下UHPLC-MS/MS检出限低,选择性好,分析时间短,更适用于检测复杂体系中极性较强和沸点较高的异噻唑啉酮类化合物。然而,UHPLC-MS/MS检测异噻唑啉酮类化合物主要集中于化妆品、纸张、胶黏剂等领域,蔬菜中异噻唑啉酮类化合物的检测尚未见报道。因此,本工作提出了一种以乙腈为提取溶剂,结合固相萃取净化技术,采用UHPLC-MS/MS测定蔬菜中4种异噻唑啉酮类化合物含量的方法,以期为蔬菜中异噻唑啉酮类化合物的检测和相关标准的制定提供技术支持。
1 试验部分
1.1 仪器与试剂
ExionLC型超高效液相色谱仪,AB SCIEX API 5500型三重四极杆质谱仪,配电喷雾离子(ESI)源;3K15型医用离心机;SP-40LN型氮气发生器;KQ-500DE型数控超声波清洗器;QSPE-24型固相萃取装置。
混合标准溶液:分别移取0.1 mL的CMIT、MIT、BIT标准储备溶液,0.005 mL的OIT标准储备溶液于100 mL容量瓶中,以10%(体积分数,下同)甲醇溶液定容,得到CMIT、MIT、BIT、OIT质量浓度分别为1,1,1,0.05 mg·L-1的混合标准溶液,现配现用。
CMIT标准物质的纯度为99.7%;MIT标准物质的纯度为100.0%±1.0%;BIT标准物质的纯度为99.8%;OIT标准物质的纯度为98.96%,于0~4 ℃避光储存;甲醇、乙腈、丙酮、二氯甲烷、乙酸乙酯均为色谱纯;其余试剂为分析纯;试验用水为超纯水。
1.2 仪器工作条件
1.2.1 色谱条件
ACQIUTY UPLC BEH SHIELD RP18色谱柱(50 mm×2.1 mm,1.7 μm);柱温 35 ℃;进样量 2 μL;流量0.3 mL·min-1;流动相A为水,B为甲醇。梯度洗脱程序:0~0.5 min时,A由0升至95%;0.5~1.5 min时,A由95%降至80%;1.5~2.5 min时,A由80%降至5%,保持1.0 min;3.5~4.0 min时,A由5%升至95%,保持1.5 min。
1.2.2 质谱条件
ESI源,正离子(ESI+)扫描模式;多反应监测(MRM)采集模式;离子喷雾电压 5 500 V;离子源温度 600 ℃;干燥气和雾化气均为高纯氮气;4种异噻唑啉酮类化合物的其他质谱参数见表1,其中“*”代表定量离子。
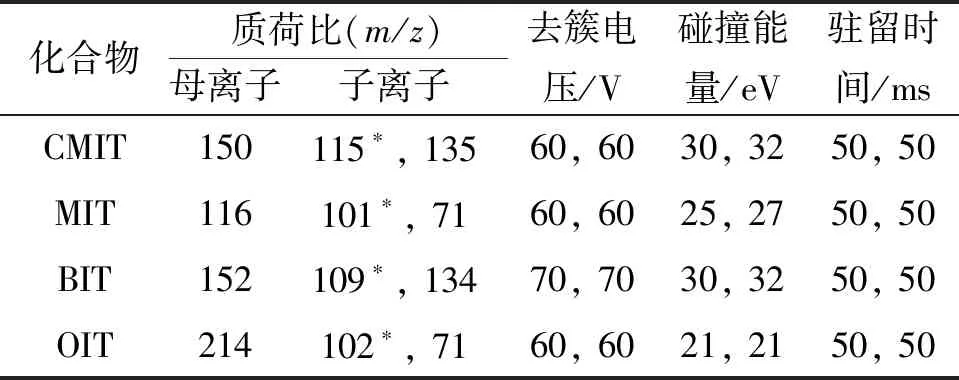
表1 质谱参数
1.3 试验方法
1.3.1 样品提取
2.3 血清因子与肺功能及EOS、ACT评分的相关性分析 血清IgE、IL-4水平与FVC 、FEV1、FEV1/FVC及ACT评分呈负相关(P<0.05),与痰EOS呈正相关(P<0.05);RPB水平与FVC 、FEV1、FEV1/FVC及ACT评分呈正相关(P<0.05),与痰EOS呈负相关(P<0.05)。见表3。
准确称取10.0 g(精确至0.01 g)已匀浆的蔬菜样品置于50 mL离心管中,加入10 mL乙腈,超声提取30 min后以转速8 000 r·min-1离心3 min。取上清液,残渣用10 mL乙腈重复提取一次,合并乙腈提取液,加入5 g氯化钠,剧烈振荡1 min后静置5 min,取上层乙腈相,于40 ℃氮吹浓缩至近干,用2 mL 10%甲醇溶液溶解残渣,待净化。
1.3.2 样品净化
将HLB固相萃取小柱置于固相萃取装置上,依次用5 mL甲醇和5 mL水活化柱子,然后将待净化的提取液全部转移到已活化平衡的HLB固相萃取小柱中,待液体全部流出,加入5 mL 10%甲醇溶液淋洗,弃去流出液,再用6 mL甲醇洗脱,收集洗脱液,于40 ℃氮吹浓缩至近干后,用甲醇定容至1 mL,过0.22 μm有机滤膜,按照仪器工作条件测定。
2 结果与讨论
2.1 色谱条件的选择
2.1.1 色谱柱
试验分别考察了SHIM-PARK CISS C18(50 mm×2.1 mm,1.9 μm)、ZORBOX ECLISE PLUS C8(50 mm×2.1 mm,1.8 μm)、Kromasil C18(50 mm×2.1 mm,1.9 μm)、ACQUITY UPLC BEH SHIELD RP18(50 mm×2.1 mm,1.7 μm)4种色谱柱对4种异噻唑啉酮类化合物的分离效果,结果见图1。
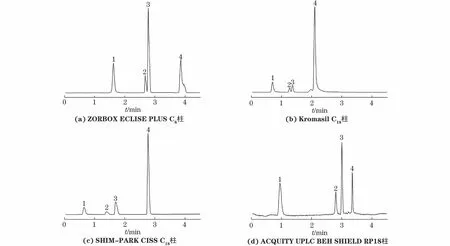
1—MIT;2—CMIT;3—BIT;4—OIT
结果表明,4种色谱柱对目标物的保留能力差别不大,但ACQUITY UPLC BEH SHIELD RP18色谱柱对目标物的分离效果相对较好,响应较高,目标物均能实现基线分离且峰形尖锐、对称。因此,试验选择ACQUITY UPLC BEH SHIELD RP18色谱柱进行分离。
2.1.2 流动相
在相同梯度洗脱条件下试验对比了分别以甲醇-水和乙腈-水为流动体系时4种异噻唑啉酮类化合物的分离效果, 结果表明, 甲醇-水体系的分离效果更好,质谱响应更高。为了增加正离子的电离程度,改善峰形,可在甲醇-水体系中加入甲酸或甲酸铵,因此试验又对比了甲醇-0.1%(体积分数)甲酸溶液体系和甲醇-5 mmol·L-1甲酸铵溶液体系对目标物的分离效果。结果显示:在流动相体系中加入甲酸后目标物的响应降低,峰形未得到明显改善;而加入甲酸铵后,可有效降低基线背景,但响应同样有所降低。为了得到更高的方法灵敏度,试验选择甲醇-水体系为流动相。
2.2 样品前处理条件的选择
2.2.1 提取溶剂
4种异噻唑啉酮类化合物都是极性化合物,易溶于水以及多种有机溶剂。选取了空白白菜基质进行加标试验,分别考察了以水、甲醇、乙腈、丙酮、乙酸乙酯作为提取溶剂时4种异噻唑啉酮类化合物的提取效果,低浓度水平下(添加CMIT、MIT、BIT质量浓度为5.0 μg·kg-1,OIT质量浓度为0.25 μg·kg-1)4种异噻唑啉酮类化合物的回收率结果见图2。
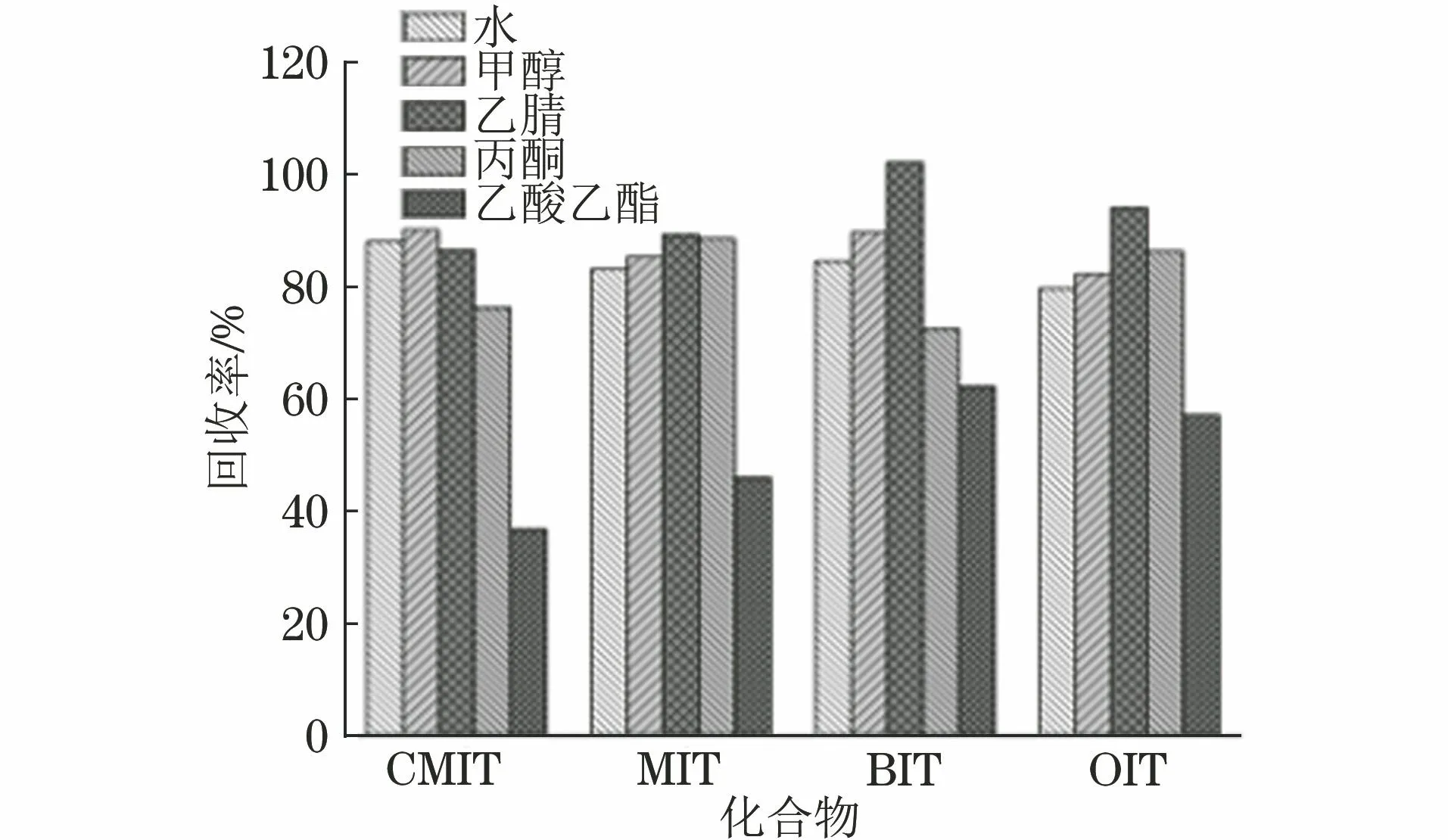
图2 低浓度水平下提取溶剂对4种异噻唑啉酮类化合物回收率的影响
结果表明:在低浓度水平下,采用乙酸乙酯提取时目标物回收效果较差;采用水、甲醇、乙腈、丙酮提取时目标物的回收率均能达到70.0%以上。考虑到随着加标浓度水平的增大,以水和甲醇提取时OIT的回收率降低,且丙酮毒性较大,试验选择乙腈作为提取溶剂。
2.2.2 固相萃取条件
蔬菜提取液中除含有大量叶绿素、叶黄素等多种色素外,还含有糖类、有机酸等杂质,基质成分十分复杂,会抑制化合物的离子化率,不利于质谱检测分析,故选择对极性化合物有很好保留效果的HLB固相萃取小柱对提取液进行净化。为了尽可能将亲水亲脂杂质均淋洗下来,需要在淋洗液中加入一定比例的有机溶剂,但有机相比例过大会将目标物一起淋洗下来,因此试验首先对淋洗液的有机相含量进行考察,分别以体积分数为1%,5%,10%,15%,20%,30%的甲醇溶液为淋洗液,考察了其对4种异噻唑啉酮类化合物回收率的影响,结果如图3所示。
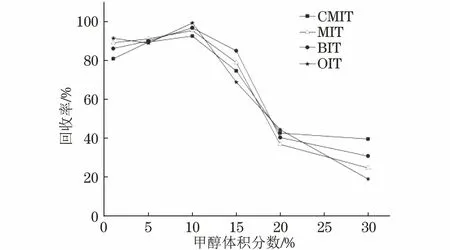
图3 淋洗液中甲醇体积分数对4种异噻唑啉酮类化合物回收率的影响
结果表明,10%甲醇溶液具有很好的淋洗效果,且淋洗流出液中未检测到目标物,故试验选择10%甲醇溶液作为淋洗液。
试验进一步考察了甲醇和乙腈两种不同极性的洗脱溶剂对目标物的洗脱效果。结果发现甲醇和乙腈对目标物的洗脱效果基本一致,考虑到甲醇沸点低于乙腈,便于后续氮吹浓缩,试验选择甲醇作为洗脱溶剂。
2.3 基质效应
基质效应是指除被测物以外样本的特征,其能够影响被测物的检测及测定结果,包括基质增强效应和基质抑制效应。基质效应(ME)=(基质溶液中目标物的峰面积/纯溶液中目标物的峰面积)×100%,ME<80%视为强基质抑制效应,80%
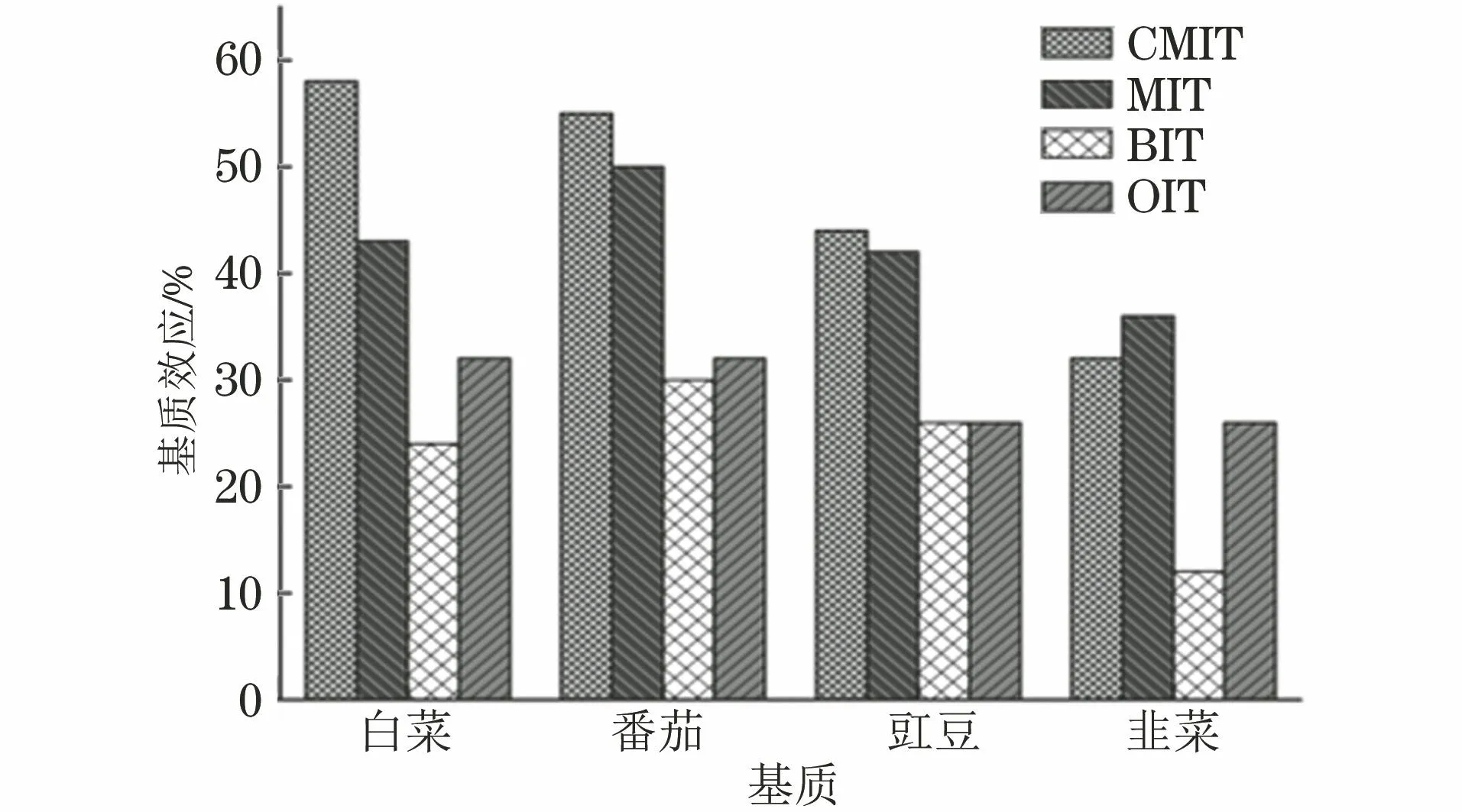
图4 4种异噻唑啉酮类化合物在不同基质中的基质效应
由图4可知,CMIT、MIT、BIT、OIT在4种蔬菜基质中均表现为强基质抑制效应,其中韭菜对目标物的基质效应整体更强,在相同蔬菜基质中BIT的基质抑制效应相对较大。因此,试验采用配制基质匹配标准溶液来绘制工作曲线,以消除基质效应。
2.4 工作曲线、检出限和测定下限
分别使用白菜、豇豆、番茄和韭菜4种蔬菜空白基质溶液配制CMIT、MIT、BIT的质量浓度为5.0,10.0,20.0,50.0,100.0,200.0,400.0,600.0 μg·L-1,OIT质量浓度为0.25,0.5,1.0,2.5,5.0,10.0,20.0,30.0 μg·L-1的混合标准溶液系列,在优化的仪器工作条件下分析,以目标物的质量浓度为横坐标,对应的峰面积为纵坐标,绘制工作曲线。结果表明,4种异噻唑啉酮类化合物的质量浓度在一定范围内与对应的峰面积呈线性关系,其线性范围、线性回归方程和相关系数见表2。
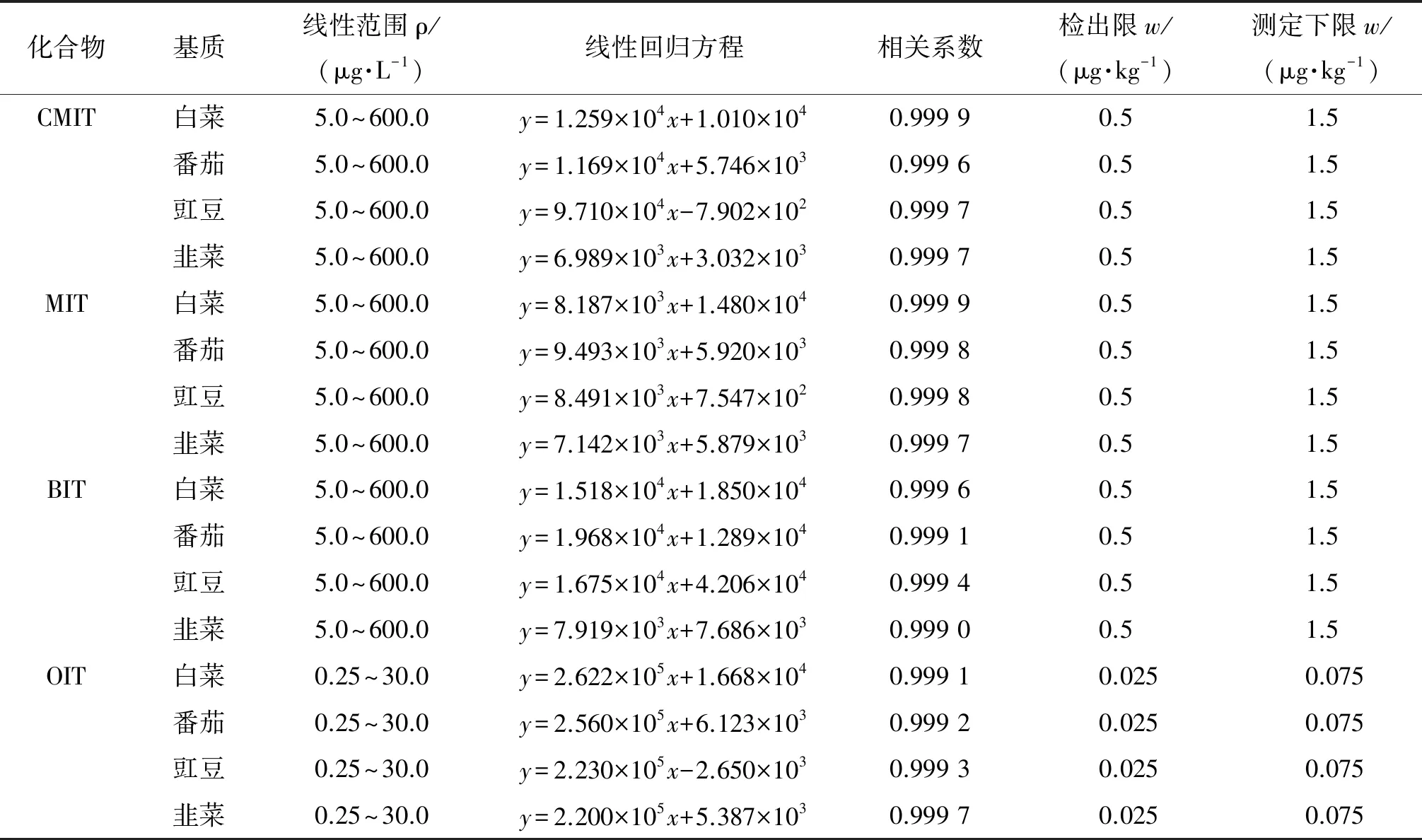
表2 线性参数、检出限和测定下限
分别采用3倍信噪比(S/N)确定方法的检出限(3S/N),10倍信噪比确定方法的测定下限(10S/N),结果见表2。
2.5 精密度和回收试验
在空白蔬菜基质溶液中加入低、中、高3种浓度水平的4种异噻唑啉酮类化合物,按试验方法进行测定,每个浓度水平平行测定6次,计算回收率和测定值的相对标准偏差(RSD),结果见表3。
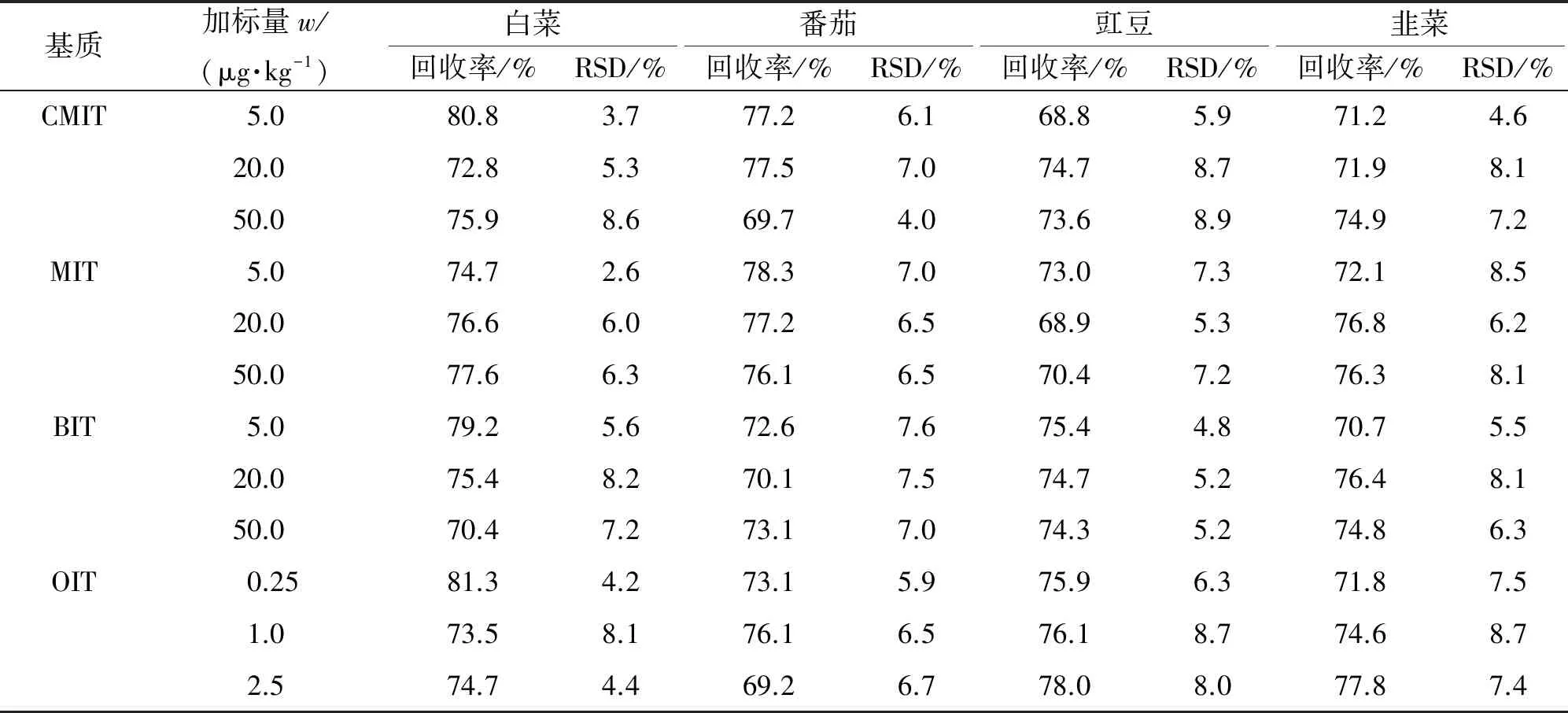
表3 精密度和回收试验结果(n=6)
由表3可知,4种异噻唑啉酮类化合物在不同蔬菜基质中的回收率为68.8%~81.3%,测定值的RSD为2.6%~8.9%,满足蔬菜中农药残留的检测要求。
2.6 样品分析
采用试验方法对市售的20种蔬菜进行测定,结果发现仅在一组韭菜样品中检出BIT,检出量为2.3 μg·kg-1,其余样品中均未检出目标物。
本工作提出了一种快速测定蔬菜中4种异噻唑啉酮类化合物含量的超高效液相色谱-串联质谱法。以乙腈为提取溶剂,经过HLB固相萃取小柱净化,采用基质匹配法定量,在优化的仪器工作条件下,能准确测定蔬菜中残留的4种异噻唑啉酮类化合物。该方法操作简单,灵敏度高,回收率、精密度均满足蔬菜中农药残留的检测要求,适用于高通量的样品检测。
猜你喜欢
煤化工(2022年3期)2022-07-08
云南化工(2021年7期)2021-12-21
天然产物研究与开发(2018年7期)2018-08-21
天然产物研究与开发(2018年4期)2018-05-07
中国资源综合利用(2016年10期)2016-01-22
中国塑料(2015年10期)2015-10-14
郑州大学学报(工学版)(2015年1期)2015-03-24
天然产物研究与开发(2014年5期)2014-04-27
中国兽药杂志(2012年4期)2012-11-06
河北大学学报(自然科学版)(2012年3期)2012-03-25
