
NIPBL基因突变致德朗热综合征临床表现和遗传学研究分析
2024-03-26 05:30朱书瑶韩彦青罗泽民
中国妇幼健康研究 2024年3期
张 衡,汤 蓓,朱书瑶,韩彦青,曾 兰,王 锦,邓 艺,陈 艾,罗泽民
(四川省妇幼保健院1.新生儿科;2.超声影像科;3.儿童健康中心;4.医学遗传与产前诊断科,四川 成都 610031)
德朗热综合征(Cornelia de Lange syndrome,CdLS)是一种罕见的显性遗传先天性畸形疾病,发病率为1/10 000~1/30 000。散发为主,少有家系报道,临床表现轻重不一,存在遗传异质性。经典型CdLS通常在胎儿期有宫内生长受限,生后有特殊颅面畸形、肢体畸形、智力障碍及生长发育落后,部分可在胎儿期及新生儿期被识别[1]。根据突变基因不同分为5个亚型(CdLS1~CdLS5),至少涉及NIPBL、SMC1A、SMC3、HDAC8、RAD21、BRD4和ANKRD11这7个基因变异。近70%的经典型CdLS患者由NIPBL基因突变引起,称为CdLS1型(OMIM 122470)。该病无特效治疗,早期易合并胃食管反流及反复感染,后期可出现口腔疾病、肥胖、关节挛缩、运动障碍等问题,需终身随访[1]。本研究通过分析我国CdLS1型患儿基因型及表现型之间的联系,以提高我国医生对该病的认识,为家系遗传及再生育提供咨询和依据。
1 资料与方法
1.1 一般资料
本研究共纳入41例CdLS1型患者,其中1例(P18)来自四川省妇幼保健院儿科个案报道,其余40例均来自文献综述。以知网、万方、PubMed数据库为文献来源,检索建库至2022年9月发表的文献。中文检索词为“德朗热,NIPBL”;英文检索词为“Cornelia de Lange,NIPBLand CHINA”。文献纳入标准:仅限我国有详细临床资料,经基因测序发现NIPBL基因突变的CdLS患者;文献排除标准:重复报道文献、基础试验、综述等。发现符合条件的英文文献5篇,中文文献20篇。本研究经四川省妇幼保健院伦理会批准,批准号:川妇幼20200424-62号。
1.2 研究方法
对41例患者的临床资料进行回顾性分析,包括性别、身高、宫内发育情况、颅面畸形、四肢畸形、智力发育、头围、头颅影像学检查、诊断年龄等,并进行临床特征性评分及基因突变类型分析。其中CdLS的临床特征评分标准依据2018年CdLS的诊断和处理专家共识[1]:①经典型CdLS:≥11分,且至少包含3项基本特征;②非经典型CdLS:9~10分,且至少包含2项基本特征;③疑似CdLS:4~8分,包含1项基本特征,需完善分子检测后诊断;④不考虑CdLS:<4分;⑤≥11分可临床诊断,见表1。

表1 CdLS的临床特征评分
1.3 统计学方法
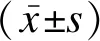
2 结果
2.1 患儿基本特征
本研究共纳入41例CdLS1型患儿,其中男16例、女23例、无性别体现2例。诊断年龄为生后胎儿期至12岁,产前诊断6.1%(2/33),新生儿期诊断30.3%(10/33)。经典型80.5%(33/41),非经典型14.6%(6/41),疑似CdLS1 4.9%(2/41)。主要临床表现为特殊颅面畸形100.0%(41/41)、肢体畸形100.0%(41/41)、智力障碍100.0%(21/21)、矮小症97.1%(33/34),见图1。其他临床表现包括先天性心脏病5例(P1、P4、P16、P20、P30)、肾囊肿3例(P8、P20、P23)、癫痫2例(P22、P33)、隐睾2例(P1、P20)、腭裂2例(P3、P16),见表2。
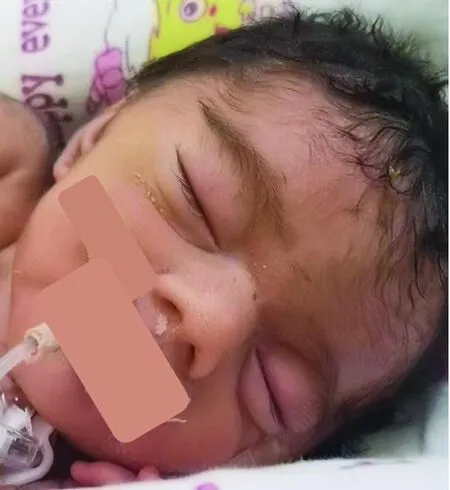
注:来源于本院患者P18(已获得监护人知情同意)。
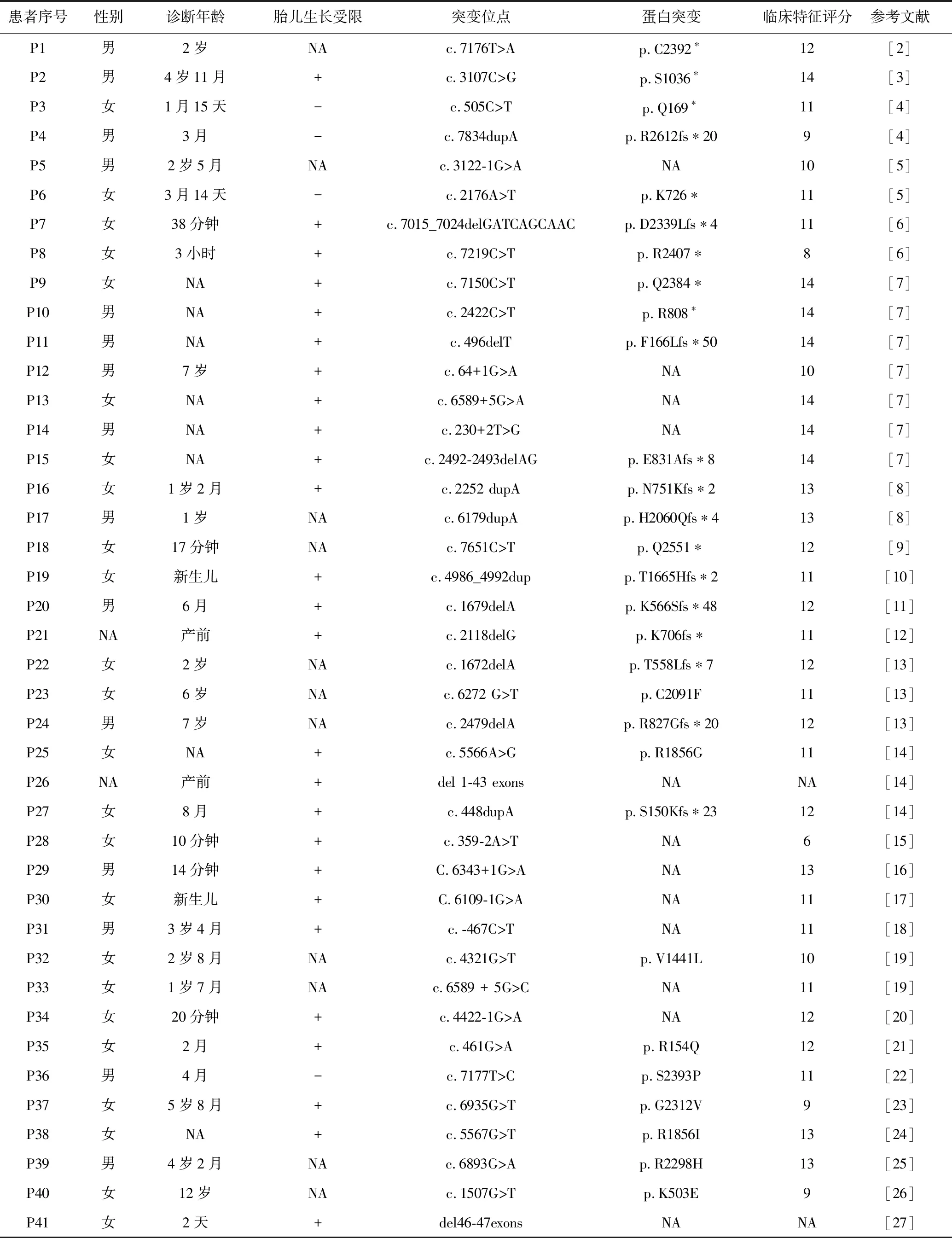
表2 我国41例NIPBL基因突变CdLS1患者资料
2.2 患儿基因变异特点
本研究41例CdLS1型患儿中,移码突变26.8%(11/41),剪切突变24.4%(10/41),错义突变22.0%(9/41),无义突变22.0%(9/41),微缺失4.9%(2/41)。新发突变97.6%(40/41),仅P31遗传于父亲,死亡1例(P12)。除P41(新生儿)和P26(引产)2例临床表现评估有限、未纳入统计分析外,其余39例CdLS1患者采用Kruskal-WallisH法分析基因型与表现型,结果差异无统计学意义(H=3.005,P=0.391),见表2。
3 讨论
3.1 CdLS的临床特点和研究进展
CdLS是一种罕见的以多系统发育障碍为主要表现的显性遗传病,在1933年由荷兰儿科医生Cornelia de Lange进行了首例报道,具有典型的颅面畸形表现:连眉,短鼻,长人中,薄上唇,其他还可出现四肢骨骼、心脏、神经系统、肾脏等发育不良。受累个体在产前及出生后均出现生长受限,可出现关节挛缩、运动发育受影响,大多伴有智力障碍。来自295例CdLS患者的大数据研究显示,婴儿早期易出现喂养困难、胃食管反流等消化道症状,严重者可因为返流导致Barrett食管炎、吸入性肺炎等,幼儿期及儿童期易出现包括肺动脉狭窄在内的先天性心脏病、呼吸道及消化道疾病,成年后心血管、呼吸、消化等系统疾病为主要死亡原因[28]。该病尚无特效治疗,需终身随访,给受累个体、家庭及社会带来极大的负担。
3.2 CdLS的致病机制
黏连蛋白复合体是一种保守的多蛋白复合物,其主要功能与染色体的准确分离有重要关联,同时在DNA修复、基因转录和调控中具有非常重要的作用。黏连蛋白复合体相关基因突变是CdLS的发病机制。蛋白功能的严重破坏会导致非整倍体和细胞死亡,其功能性缺失可导致发育缺陷。2004年Tonkin.E等人首先发现CdLS与NIPBL基因突变有关,随后8年内相继发现黏连蛋白复合体其他亚基相关的基因突变会导致CdLS。Kline[1]等人2018年发表了CdLS诊断和处理的专家共识,共识提出CdLS的诊断应该同时结合临床特征与分子遗传学检测。考虑到分子遗传检测水平在各地区存在差异,典型表现的CdLS在新生儿期易被有经验的儿科医师及遗传学医师所发现,共识指出CdLS临床诊断评分≥11分,即使没有分子遗传学检测也可临床诊断为CdLS。
3.3 CdLS基因分类特征
根据基因突变不同分为5个亚型(CdLS1~CdLS5),CdLS1型最为常见约占全部病例的70%,由NIPBL基因突变所致,呈常染色体显性遗传;CdLS2型与SMC1A基因突变相关,2006年发现为X连锁显性遗传,约占全部病例的5%左右;2012年发现的X连锁显性遗传CdLS5型由HDAC8基因突变所致,约占5%左右;2007年和2012年分别发现CdLS3型由SMC3或RAD21基因突变所致,约占全部病例的1%[29]。我国与国外研究一致,以NIPBL基因突变为主,偶有HDAC8基因突变等报道。
NIPBL基因位于染色体5p13.2,在脊椎动物中高度保守,NIPBL基因与小鼠同源性达96%[30],编码一个包含2 804个氨基酸的蛋白,NIPBL蛋白与MAU2蛋白形成异二聚体复合物,加载黏连蛋白复合体到DNA上,并使其相结合(图2)。NIPBL基因是一种黏连蛋白调控基因,在组织中广泛表达。NIPBL基因突变导致胚胎发育异常,在果蝇、小鼠、斑马鱼等动物试验中都被验证。
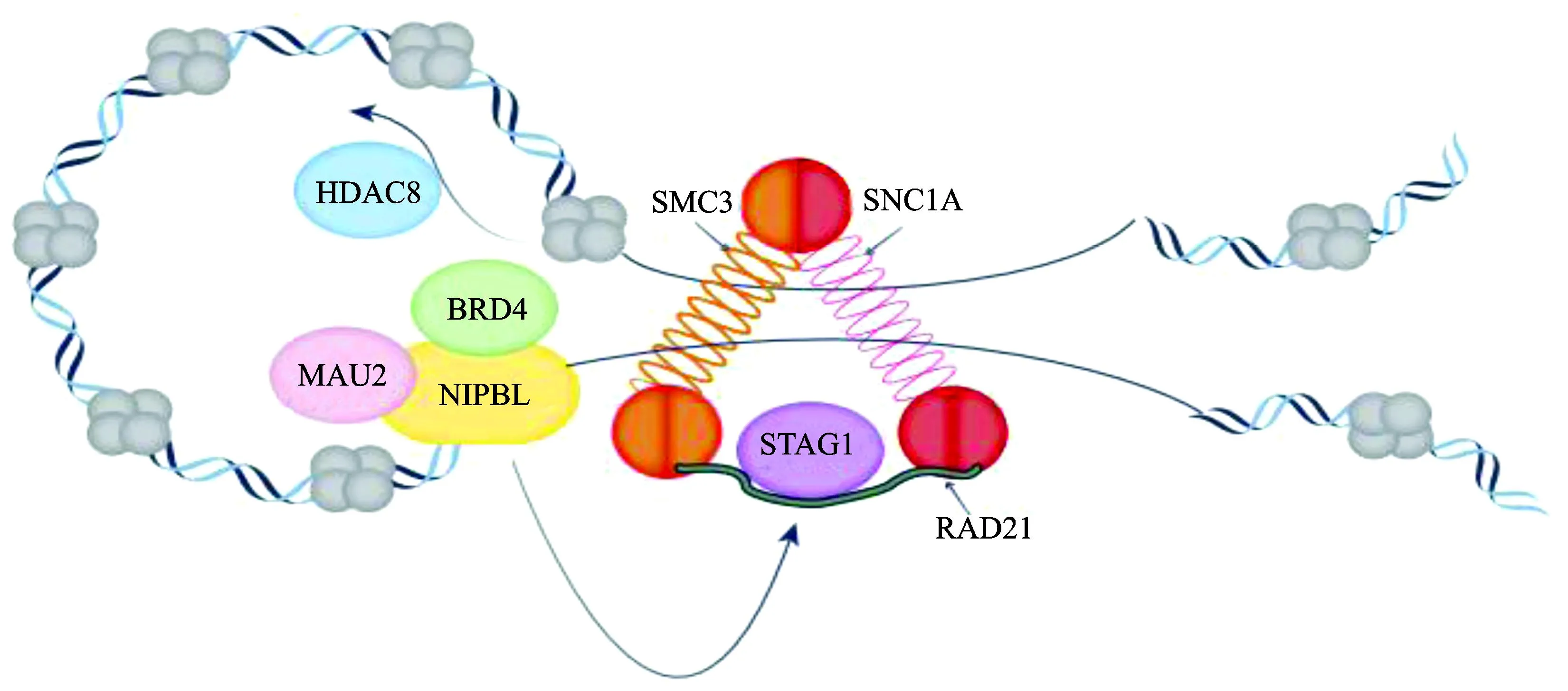
图2 部分黏连蛋白复合体及其调控因子在染色体分离中的结构和作用
3.4 治疗与产前诊断
本研究通过对39例病例统计分析,发现突变类型与临床特征并无相关性,与Tonkin E等于2004年研究发现截短突变患者表型较错义突变更为严重存在差异。考虑原因可能为①本研究样本量较小,多因素分析受限;②本研究多为经典型患者,临床表现差异小;③评分为回顾性分析存在描述差异。值得注意的是,不建议通过临床表现划分疾病严重程度,而是根据遗传病因进行分类[1]。先天性膈疝是CdLS的基本特征及婴儿期死亡原因之一,但本研究统计中的CdLS病人并无先天性膈疝报道。
目前对CdLS尚无特效的治疗措施,仅能对症治疗,由于涉及多个组织,需多学科长期共同协作。近期有研究发现,通过调节与NIPBL蛋白功能相反WAPL基因编码的粘黏蛋白释放因子含量,使粘连蛋白复合体与DNA转载水平达到动态平衡的状态可减轻NIPBL基因敲出小鼠胚胎期的畸形程度[30],这为CdLS患者以后的基因治疗提供了一个方向。
总之,约80%CdLS患者在胎儿期产前检查时可出现宫内生长受限,66%出现肢体异常,50%有胎儿面部异常轮廓,建议对这类胎儿应进行CdLS筛查,以尽早明确诊断。如何利用特定的形态学异常提高产前诊断,是我们接下来需要思考的,也为功能影像科、产前诊断医生带来了挑战。
猜你喜欢
英语世界(2023年6期)2023-06-30
小学生作文(低年级适用)(2022年10期)2022-10-31
中学生数理化·七年级数学人教版(2022年11期)2022-02-14
现代临床医学(2021年1期)2021-01-26
中国生殖健康(2020年2期)2021-01-18
小学生导刊(2018年13期)2018-06-29
原子与分子物理学报(2015年1期)2015-11-24
河南医学研究(2014年5期)2014-02-27
食品科学(2013年15期)2013-03-11
地球学报(2012年1期)2012-09-20
