
MAP3K7、MPO基因与中国人群坏疽性脓皮病发病的相关性研究
2024-03-26 06:32:42靳传洋王真真孙乐乐刘亭亭张福仁
中国麻风皮肤病杂志 2024年4期
靳传洋 王真真 孙乐乐 刘亭亭 刘 红 张福仁
1山东第一医科大学附属皮肤病医院,山东济南,250022;2 山东省皮肤病性病防治研究所,山东济南,250022
坏疽性脓皮病(pyoderma gangrenosum, PG)是一种以皮肤炎症和溃疡为主要表现的非感染性、自身炎性、嗜中性皮肤病。PG表现为快速发展的疼痛性皮肤溃疡,常常发生在下肢和躯干[1]。其发病率在美国成年人中为58/107,在英国人口中约为6/107[2]。其病因尚不明确,目前观点认为,该病是在遗传倾向的背景下,先天和适应性免疫系统失调造成的[3]。近年来的研究发现了10个与PG发病相关的基因:NLRP3,MEFV,NOD2,LPIN2,PSMB8,NLRP12,PSTPIP1,PTPN6,JAK2和NFκB1[4-8]。这些基因突变后主要是通过影响NF-κB通路来影响多种细胞因子的过度表达(如IL-1α,IL-1β,IL-36,G-CSF等),进而影响中性粒细胞的聚集,延长中性粒细胞寿命,从而导致PG的发病[2]。
既往对PG易感基因的报道多集中在病例报道和小样本的研究中(最大样本量为14例PG患者和20例对照)[6],且研究的种群多集中在欧美人种,仅有1例对中国汉族群体的病例报道[9]。因此,我们汇总了一组可能与PG发病相关的基因,囊括了既往报道的10个基因(NLRP3,MEFV,NOD2,LPIN2,PSMB8,NLRP12,PSTPIP1,PTPN6,JAK2和NFκB1);同时,在对既往报道的PG相关基因PTPN6所在炎症通路的研究中发现,MAP3K7,MyD88,SYK,RIRPK1在促进IL-1α、TNF、G-CSF等细胞因子分泌起关键作用[10]。此外,PG患者中存在髓过氧化物酶(myeloperoxidase, MPO)功能缺陷的报告[11]提示:髓过氧化物酶编码基因MPO有害性突变可能是PG患者中髓过氧化物酶功能缺陷的原因。因此,我们也纳入了PTPN6所在炎性通路中的4个相关基因(MAP3K7,MyD88,SYK,RIRPK1)和髓过氧化物酶编码基因MPO,共计15个基因,对36例中国汉族PG患者的此组基因进行目标基因二代测序(next generation sequencing,NGS),旨在探索该病的遗传学背景与种族差异性。
1 资料与方法
1.1 临床资料 选取2015-2022年来我院就诊的36例坏疽性脓皮病散发患者,年龄10~89岁,男22例,女14例,平均年龄56.06岁,平均发病年龄54.69岁。经过我院皮肤病医师临床诊断及组织病理学诊断确诊为坏疽性脓皮病。448例对照采用全外显子测序,测序结果储存于山东第一医科大学附属皮肤病医院数据库,对照组男356例,女92例,平均年龄69.67岁。该研究通过山东第一医科大学附属皮肤病医院伦理委员会审批。
1.2 方法
1.2.1 DNA提取 从-80℃超低温冰箱取出外周血,使用TIANamp Blood DNA Kit DP318-02(天根血液基因组DNA提取试剂盒),TGudieM16 全自动核酸提取仪提取外周血基因组DNA。
1.2.2 目标区域测序及注释 使用Access Array公司微控流技术对36例坏疽性脓皮病患者的15个基因进行目标区域测序。15个基因包括既往文献中报道的10个突变基因NLRP3,MEFV,NOD2,LPIN2,PSMB8,NLRP12,PSTPIP1,PTPN6,JAK2,NFκB1以及可能与PG发病相关的基因共5个:SYK,MYD88,RIPK1,MAP3K7,MPO。测序结果基于hg19参考基因组,用ANNOVA软件对所有36例PG患者与448例全外显子测序数据中的此15个基因中存在的变异位点进行注释。除去千人基因计划数据库2015版与ExAC数据库中等位基因频率大于0.005的数据后,满足以下任意一条即认为该突变为有害突变:(1)stop gain, stoploss, frameshift, splicing;(2)满足 SIFT_score<0.05, Polyphen2_HDIV_score >=0.957, CADD_pred>15 并且排除ACMG 2015标准中“Benign”和“Likely benign”的突变位点。注释后再进行显性遗传模式和隐性遗传模式的基因负荷检验。
1.2.3 Sanger测序 我们对值得讨论的致病性突变位点进行了Sanger测序验证。使用NCBI网站的Primer-BLAST引物设计工具进行引物设计,分别获取外显子的跨侧翼的上下游引物序列,由华大基因公司合成。PCR反应在美国ABI公司9700 型PCR扩增仪上完成,使用醋酸钠乙醇沉淀法纯化PCR产物。最后在美国ABI公司3500XL遗传分析仪直接测序。
2 结果
2.1 NGS测序结果 按照我们的有害性筛选标准,在其中4例患者中的5个基因中,发现了6个有害性突变位点。包括一个移码突变: NLRP12:c.908delT(p.F303fs)和 5个错义突变:MAP3K7: c.815C>G(p.S272C), MPO: c.1484C>T(p.T495I), MPO: c.1705C>T(p.R569W), NOD2: c.2975G>A(p.R992Q), SYK: c.389A>G(p.E130G) (MAP3K7, MPO, NOD2, SYK 和NLRP12 的氨基酸序列分别基于NM_003188, NM_000250, NM_001293557, NM_001135052 和NM_001277126转录本)。经过显性遗传模式和隐性遗传模式的基因负荷检验,我们未发现有统计学差异的突变基因(表1)。然而我们发现了两个值得讨论的突变。MAP3K7的杂合突变和MPO的纯合(复合杂合)突变,此两种突变仅存在于病例组,提示:MAP3K7与MPO可能是PG发病的关联基因。
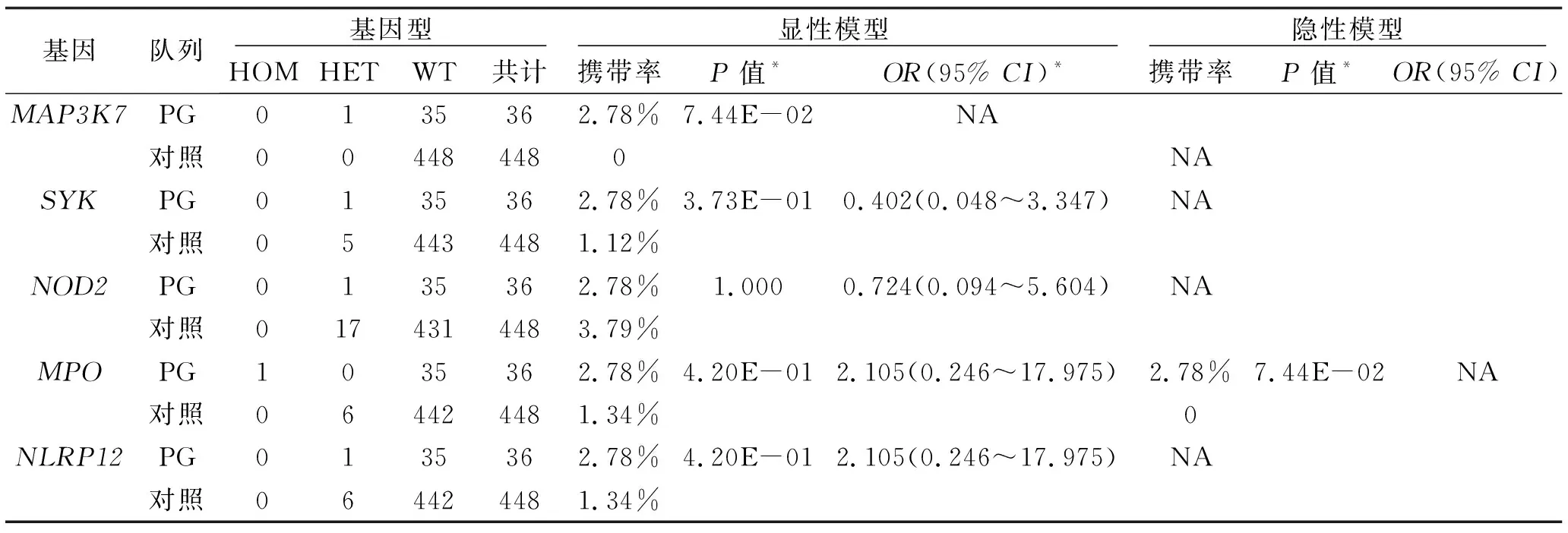
表1 基因负荷检验结果
2.2 Sanger测序结果 我们对3个值得讨论的致病性突变位点进行了Sanger测序验证,排除了假阳性。测序结果如图1。
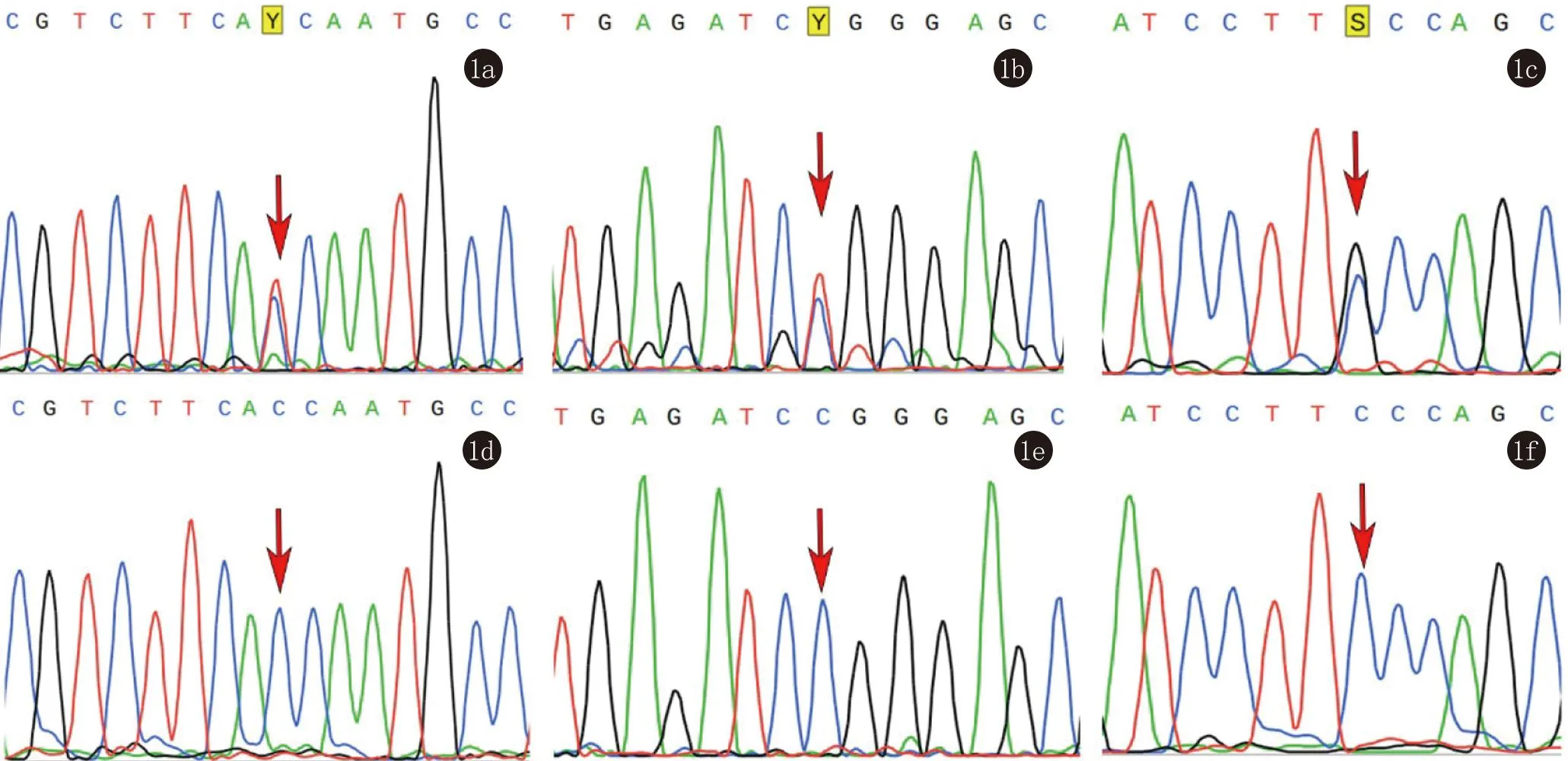
1a:MPO错义突变: c.C1484T in exon 9;1b:MPO错义突变: c.C1705T in exon 10;1c:MAP3K7错义突变:c.C815G in exon 8;1d:MPO正常对照:c.1484C in exon 9;1e:MPO正常对照: c.1705C in exon 10;1f:MAP3K7正常对照:c.815C in exon 8图1 Sanger测序结果
3 讨论
在我们的病例样本中,共在MAP3K7,SYK,NOD2,MPO,NLRP12基因中发现了致病性突变位点,其中MAP3K7的杂合突变和MPO纯合(复合杂合)突变引起了我们的关注,这两个基因突变仅见于PG患者群体中,在448例对照中均无出现。MAP3K7基因编码TAK1(tumor growth factor-β activated kinase 1),Prajwal Gurung团队通过对Ptpn6spin小鼠的研究发现TAK1是引发该种小鼠嗜中性粒细胞疾病的关键。Ptpn6spin小鼠是PTPN6基因纯合突变模型,导致SHP1上208位氨基酸Tyr突变为Asn,该种小鼠会出现持续性的足垫肿胀和化脓性炎症,与人类嗜中性皮肤病非常相似[12]。在对该种小鼠的研究中发现:脾酪氨酸激酶 (spleen tyrosine kinase,SYK)是一种可磷酸化MyD88(myeloid differentiation primary response gene 88)的关键激酶,MyD88磷酸化后可将信号转导到RIRK1(receptor interacting protein kinase 1,由RIRPK1基因编码)和TAK1(tumor growth factor-β activated kinase 1 ,由MAP3K7基因编码),最终促进 IL-1α,TNF,G-CSF等细胞因子分泌,并在PTPN6spin小鼠中介导皮肤病;RIPK1支架功能和TAK1信号传导在Ptpn6spin小鼠足垫炎症中发挥重要作用,我们在人类PG患者中发现的MAP3K7突变,提示RIPK1-TAK1信号轴可能在人类PG发病过程中也发挥着相似的作用。
此外,我们发现的MPO复合杂合突变(c.1484C> T;c.1705C>T)是队列中纯合(复合杂合)状态下观察到的唯一变化。该患者具有的c.1705C>T错义突变在先前髓过氧化物酶缺乏症(MPOD)患者中曾被报道过[13]。 髓过氧化物酶是中性粒细胞嗜天青颗粒和单核细胞溶酶体中的重要成分[14],髓过氧化物酶缺乏症是一种常染色体隐性遗传病,在1954年首次描述,该病是吞噬细胞常见的遗传缺陷,表现为对病原微生物杀伤能力受损[15]。MPOD患者可伴发嗜中性皮肤病,例如全身泛发性脓疱型银屑病(Generalized pustular psoriasis,GPP)[13]和PG[11]。GPP是一种无菌性脓疱性疾病,可以表现为急性的或持续性进行性发作,有时可有危及生命的多系统炎症发作。我们在该例MPO复合杂合突变的PG患者中,发现中性粒细胞计数[9.89×109,参考值(1.8~6.3)×109]增加和中性粒细胞比例(82.4%,参考值40%~55%)增加的现象,这一现象在MPO突变的GPP患者中也有出现。
MPO功能丧失突变导致GPP发病的机制,已经有了相对清晰的阐释。Haskamp等证明,GPP患者中的MPO功能丧失突变导致中性粒细胞和单核细胞中的MPO缺乏,并且导致中性粒细胞中中性粒细胞丝氨酸组织蛋白酶(neutrophil serine proteases cathepsin G,CTSG)的活性增加。CTSG的活性增加会导致IL-36前体向IL-36的转化增加。其次,MPO缺陷导致中性粒细胞细胞外陷阱(neutrophil extracellular trap,NET)的形成减少,导致与NET结合蛋白酶减少,激活IL-36前体的可溶性中性粒细胞蛋白酶占优势。此外,在MPO缺陷患者和MPO-/-小鼠中,单核细胞对中性粒细胞的吞噬作用(中性粒细胞胞葬作用)受损,这表现为中性粒细胞在皮肤炎症部位的抵抗时间延长和外周血中性粒细胞计数增多[13]。这些发现表明,MPO在皮肤中充当神经营养因子相关炎症的调节剂,MPO的功能丧失突变通过IL-36高活化和中性粒细胞胞葬作用受损诱导脓疱型银屑病的发生[16]。IL-36也是一种被认为在PG病理生理学中起重要作用的细胞因子[17],在PG患者的皮损中亦可观察到密集的中性粒细胞浸润和NET聚集[18],在我们MPO复合杂合突变的患者中也发现了中性粒细胞计数增加的现象。这些提示,MPO功能丧失突变在PG的发病过程中,可能发挥着与在GPP发病过程中相似的作用。
总之,我们对既往研究的可能与坏疽性脓皮病15个基因进行了目标区域测序,在MAP3K7,SYK,NOD2,MPO,NLRP12基因中发现了功能丢失突变位点,其中NOD2与NLRP12基因突变在一项针对意大利PG患者的研究中被报道过[4]。我们在中国汉族的PG患者中发现了这两个基因的突变,表明坏疽性脓皮病中存在的NOD2,NLRP12基因的突变可能是跨越种族的。此外,我们发现了MAP3K7,MPO基因功能丢失突变可能与坏疽性脓皮病的发病相关。其中,MAP3K7在Ptpn6spin小鼠发生类似人类嗜中性皮肤病的过程中起着关键作用,PTPN6突变在美国坏疽性脓皮病患者中被报道过[5],MAP3K7基因上功能丢失的突变,提示了RIPK1-TAK1信号轴可能在PG发病过程中起着重要作用。早在1991年,Disdier等就发表过一例法国PG患者中存在髓过氧化物酶缺陷的病例报道[11],MPO突变可能是髓过氧化物酶缺陷的原因,这提示MPO与PG的发病可能有着密切的关系。
我们的研究也存在着不足,这可能是由于本研究的样本量太小而没有发现显著性差异,需要后续更大样本量的探索。总之我们的研究发现了两个有提示意义的基因:MPO与MAP3K7,证明了PG是一种复杂的多基因相关自身免疫性疾病,为进一步探明PG发病机制提供了遗传学的视角。
猜你喜欢
黑龙江大学自然科学学报(2022年4期)2022-11-17 08:07:52
河北科技师范学院学报(2022年2期)2022-08-26 08:55:46
种子(2021年3期)2021-04-12 01:42:22
中华养生保健(2020年3期)2020-11-16 00:53:10
中国中西医结合皮肤性病学杂志(2016年4期)2016-07-18 10:59:56
中国塑料(2016年3期)2016-06-15 20:30:00
外语教学理论与实践(2016年1期)2016-06-11 05:51:48
实用皮肤病学杂志(2015年4期)2015-12-22 11:21:41
华东理工大学学报(自然科学版)(2015年4期)2015-12-01 04:00:36
河南医学研究(2014年7期)2014-02-27 14:53:27
