
常染色体隐性遗传性羊毛状发伴少毛症两家系基因突变分析
2024-03-26 01:52:28赵安琪曹巧玉郑璐瑶刘庆梅吴文育赵敬军
中国麻风皮肤病杂志 2024年4期
赵安琪 曹巧玉 郑璐瑶 刘庆梅 李 明 吴文育 赵敬军
1上海交通大学医学院附属新华医院皮肤科,上海,200092;2上海交通大学医学院皮肤病研究所,上海,200092;3复旦大学附属儿科医院皮肤科,上海,201102;4安徽省儿童医院皮肤科,安徽合肥,230022;5复旦大学附属华山医院皮肤科,上海,200040
遗传性少毛症是一组相对罕见的遗传性皮肤病,具有广泛的基因谱和表型谱,根据是否伴随其他系统损害分为综合征型和非综合征型。非综合征型遗传性少毛症仅表现为毛发的异常,其毛发表型包括先天性少毛/无毛、羊毛状发、念珠状发等。目前已有文献报道了两种遗传模式,分别是常染色体显性遗传和常染色体隐性遗传[1]。在非综合征型遗传性少毛症中,常染色体隐性遗传性羊毛状发伴少毛症(autosomal recessive woolly hair/ hypotrichosis, ARWH/HT,OMIM 278150/604379)是一种发生于高加索人或亚洲人群中的遗传性疾病。患者通常在出生时即表现出毛发的异常特征,其头发呈粗糙、无光泽、干燥、卷曲,形成发干螺旋状或波浪卷曲的外观,导致头发呈现弥漫性羊毛状,并伴有不同程度的毛发稀疏。患者的毛发生长缓慢,到达一定长度后即停止生长,同时可能伴有毛发色素减退。眉毛、睫毛、胡须、腋毛和阴毛可能呈现稀少或正常状态。其他方面,如牙齿、指(趾)甲及汗腺的发育正常,极少数患者可能伴有掌跖角化症和毛周角化症。目前已报道的致病基因包括:LIPH,LPAR6,C3ORF52,KRT25[2]。我们收集了两例ARWH/HT家系,并进行了突变检测以进一步了解该疾病的遗传基础。
1 病例资料
先证者1,男,24岁。出生后出现头发卷曲、稀少,头发生长缓慢,至2~3 cm后停止生长,易扯断,眉毛、睫毛、腋毛、阴毛稀疏,牙齿、指甲、皮肤无明显异常。父母非近亲结婚,家族成员中无类似疾病患者。先证者2,女,7岁,表现为生后毛发卷曲、稀疏,头发生长至3~4 cm后停止生长,发质粗糙,易缠绕打结,易折断,牙齿、指甲、皮肤无明显异常。父母非近亲结婚,家族成员中无类似疾病患者。
体检:两例患者一般情况可,营养、智力、骨骼均发育正常,心、肺等系统检查未见异常。皮肤科检查:先证者1头发稀疏明显,可见头皮裸露,头顶部、后枕部可见头发整体卷曲,发质粗糙,卷曲缠绕成团。眉毛、睫毛、腋毛和阴毛较稀疏(图1a)。先证者2头发卷曲呈羊毛状改变,稍稀疏,眉毛、睫毛、腋毛和阴毛较稀疏,患者均无牙齿、黏膜、指甲等其他外胚层发育异常表现(图1b)。综合上述病史和体检结果,诊断为羊毛状发伴少毛。
2 基因突变检测
2.1 外周血基因组DNA提取 经上海交通大学医学院附属新华医院伦理委员会批准(批件号XHEC-D-2019-115),以及先证者1本人和先证者2的父母签署知情同意书后,分别采取先证者及其父母外周血2 mL,应用QIAamp DNA Blood MinikitⅠ(德国QIAGEN公司)试剂盒提取全血基因组DNA。同样方法提取100例无亲缘关系的健康个体血样基因组DNA作为对照。
2.2 二代靶向测序和Sanger测序 使用二代遗传性皮肤病靶向测序包(上海安百隆生物科技有限公司)检测先证者的突变基因,该测序包纳入554个与遗传性皮肤病密切相关的基因,靶向测序的范围涵盖目标基因外显子及毗邻剪接区域约20 bp。设计探针由遗传性皮肤病权威专家设计,由瑞士罗氏NimbleGen合成。测序平台选用IlluminaHiseq X Ten高通量测序仪测序,平均测序深度可达200×以上。在相关变异频率数据库[寡核苷酸多态性数据库(dbSNP)、千人基因组数据库、ClinVar数据库和人类基因突变数据库(HGMD)]中对致病变异位点进行评估,根据美国医学遗传学与基因组学学会(ACMG)遗传变异分类指南及患者的临床表型进行致病变异的筛选。使用Primer-BLAST工具(https://www.ncbi.nlm.nih.gov/)设计基因引物,并通过PCR扩增,用ABI3730XL型全自动测序仪进行Sanger测序。E5正向引物5’-ATTAGCCTTGCAGTGGATGAGTG-3’,反向引物5’-CCTGCCAGGTATAAGCAGGAAT-3’,扩增片段长度为407 bp;E6正向引物5’-GGGTCTTTCACCAAAGCATA-3’,反向引物3’-GAGGAAACTGGTTAGAC-5’,扩增片段长度为434 bp;E7正向引物5’-GATTGGATCAACCTGGCTGC-3’,反向引物5’-ACATGCAGAATGGGCTCTCC-3’,扩增片段长度为306 bp;反应条件:96℃预变性3 min,96℃变性30 s,在相应Tm值温度退火30 s,72℃延伸30 s,循环35次,73℃充分延伸5 min,4℃保存。
2.3 剪切突变验证 对于先证者携带的剪切突变,我们进行了Minigene实验的验证。首先,将目的基因片段通过质粒转染到293T细胞系中,进行细胞培养。随后,采用Trizol法提取培养细胞中的RNA,并进行RT-PCR实验,设计特定于突变位点的PCR引物,针对剪切突变c.982+12A>G设计引物序列为:LIPH-E7F:AGTGATGGTGAGTGTGGTTGTTGCTTTTGCCT-AA;LIPH-E7R:AACCACACTCACCACTCACTGCAGAATG。针对剪切突变c.629-1_629delinsTT设计引物序列为:LIPH-E5F:TATTACTGGGCTACAAGGAGCCAT ;LIPH-E5R:CTCCTTGTAGCCCAGTAATAAAAGAGAACACATCTGCT。对PCR产物进行凝胶电泳验证后,我们回收产物并进行Sanger测序,以验证其剪切方式。
3 结果
3.1 二代皮肤靶向测序包筛选和Sanger测序验证结果 两例先证者均检测到了LIPH的复合杂合突变。先证者1的LIPH第6号外显子第742位核苷酸由C突变为A(c.742C>A),导致第248位氨基酸由组氨酸变为天冬氨酸(p.His248Asn),同时第7号外显子下游第12位核苷酸由A突变为G(c.982+12A>G),Sanger测序验证显示先证者父亲为c.982+12A>G杂合突变携带者,先证者母亲为c.742C>A杂合突变携带者。家系图和突变携带情况见图2a、2b。

2a、2b:家系1的先证者和父母突变信息;2c、2d:家系2的先证者和父母突变信息图2 家系图和先证者及父母的突变信息
先证者2的LIPH第5号外显子上游第1位至编码区第629位核苷酸由GC突变为TT(c.629-1_629delinsTT),同时第5号外显子编码区第687和688位核苷酸之间插入17 bp (c.686delinsGTAGAAC-CCAACCTGGCT),该插入突变会导致从229位天冬氨酸开始发生移码突变,移码37位氨基酸后编码终止(p.Asp229GLyfs*37),Sanger测序验证显示先证者父亲为c.686delinsGTAGAACCCAACCTGGCT杂合突变携带者,先证者母亲为c.629-1_629delinsTT杂合突变携带者。家系图和突变携带情况见图2c、2d。100例对照中均未检测到该四种突变。SIFT和Polyphen-2软件预测p.His248Asn和p.Ile220Argfs*29均为有害变异位点,可能影响蛋白质功能。
3.2 剪切突变验证结果LIPH剪切突变c.982+12A>G是指第7号外显子下游第12位核苷酸由A突变为G,突变示意图如图3a中黄色标注所示。Minigene验证发现该剪切突变会使剪切位点后延,导致7bp内含子序列滞留,图3b展示了Sanger测序结果和错误剪切,内含子滞留部分标红。
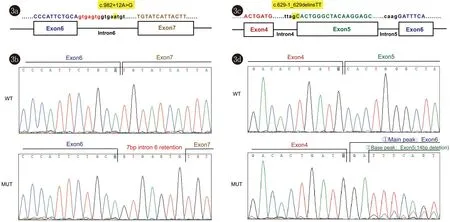
3a:LIPH c.982+12A>G剪切突变示意图,如黄色标注所示;3b:Sanger测序结果,该剪切突变导致7 bp内含子序列滞留(标红);3c:LIPH c.629-1_629delinsTT剪切突变示意图;3d:Sanger测序结果,两种错误剪切:①突变所在5号外显子被跳过(主峰);②5号外显子前AG缺失后,将内部AG识别为剪切信号,导致缺失14 bp(底峰)图3 剪切突变验证结果
LIPH剪切突变c.629-1_629delinsTT是指第5号外显子上游第1位至编码区第629位核苷酸由GC突变为TT,突变示意图如图3c中黄色标注所示。Minigene验证发现该剪切突变会导致复杂剪切变异,同时存在两种错误剪切:①突变所在5号外显子被跳过(主峰);②5号外显子前AG缺失后,将内部13-14位的AG碱基识别为剪切信号,导致缺失14 bp(底峰);这两种错误剪切如图3d所示。
4 讨论
常染色体隐性羊毛状发/少毛症(ARWH)是一种罕见的遗传性毛发疾病,临床特征为出生时或生后早期出现头发卷曲稀疏。目前已报道的基因包括LIPH,LPAR6,KRT25和C3ORF52等[2]。LIPH基因定位于人染色体3q27上,含有10个外显子,编码451个氨基酸,它编码脂肪酶H,也称为膜相关性磷脂酸-选择性磷脂酶A1α,主要功能是催化水解磷脂酸产生2-酰基溶血磷脂酸(2-acyl lysophosphatidic acid, LPA)。C3ORF52能与LIPH相互作用,影响其催化功能[3]。LPA会激活溶血磷脂酸受体6(lysophosphatidic acid receptor 6,LPAR6),LIPH、LPAR6或C3ORF52的功能丧失会导致LIPH- LPA -LPAR6信号传导减少,导致表皮生长因子受体信号传导的反激活减少,从而影响毛囊发育和毛发生长[2]。目前已报道的LIPH基因致病突变位点有34个,包括12个错义/无义突变,4个剪接突变,7个缺失突变,4个插入突变,以及4个缺失-插入突变(www.hgmd.cf.ac.uk)。
ARWH具有遗传异质性,因种族和国家/地理区域而异。在巴基斯坦ARWH人群中,47.2%由LIPH突变导致,其余52.8% ARWH患者由LPAR6突变导致。在巴基斯坦LIPH突变患者中,超过一半携带c.659_660delTA (p.Ile220Argfs*29)突变。在日本ARWH人群中,98.7%携带LIPH突变,且热点突变为c.736T>A (p.Cys246Ser)和c.742C>A (p.His248Asn)[2]。在我国尚未有统计LIPH突变占比和热点突变情况。据我们统计,目前中国已经报道的LIPH突变患者中,c.736T>A和c.742C>A突变占75%左右[4-12]。
目前,已有日本的两项研究发现,外用米诺地尔可以改善LIPH突变导致的ARWH患者。在其中一项前瞻性非随机临床试验中,共纳入8例携带LIPH致病突变的ARWH患者和1例TRPS1致病突变导致的毛发-鼻-指综合征患者,所有纳入者均每天2次外用1%米诺地尔,持续一年,发现所有具有LIPH致病变异的患者少毛症状均有改善,毛发-鼻-指综合征患者的少毛症没有改善[13]。在另一项研究中,共纳入4例具有LIPH突变的日本ARWH患者,所有患者在局部应用米诺地尔后均出现毛发生长[14]。
我们团队在2020年报道一例羊毛状发患者携带LIPHc.686delAinsGTAGAACCCAACCTGGCT突变,该突变与先证者2的突变位点一致,导致D229Gfs*37的移码突变,最终产生截断蛋白[6]。此外,本报道中发现了两个新的LIPH剪切突变c.982+12A>G和c.629-1_629delinsTT。剪切突变是指影响基因剪切过程的一类突变,可能引起多种转录本的变化,包括外显子跳跃、内含子保留、部分外显子缺失以及复杂剪切变异(包括外显子跳跃、内含子保留等多种组合)等。而这些异常剪切的结果是蛋白质缺失一长段氨基酸或者更为常见的翻译提前终止(插入、缺失的序列往往不是3的倍数)。我们通过Minigene实验验证发现该剪切突变导致的错误剪切。其中,c.982+12A>G导致出现7 bp的内含子滞留,c.629-1_629delinsTT则同时导致了5号外显子跳跃和部分5号外显子缺失。我们的研究进一步丰富了LIPH的基因谱,具体机制有待进一步功能学实验证明。
猜你喜欢
中国现代医生(2022年19期)2022-11-04 10:13:29
临床输血与检验(2022年3期)2022-06-22 02:52:50
昆明医科大学学报(2022年4期)2022-05-23 13:04:50
基层中医药(2021年8期)2021-11-02 06:24:54
郑州大学学报(医学版)(2019年3期)2019-06-03 06:19:32
传染病信息(2019年2期)2019-05-17 13:16:04
中国生殖健康(2019年11期)2019-01-07 01:27:32
青少年科技博览(中学版)(2017年5期)2018-02-28 21:23:59
中国医疗美容(2015年2期)2015-07-19 10:11:59
重庆医学(2015年12期)2015-03-05 05:52:54
