
结节性硬化症5个家系致病变异的鉴定
2024-03-22 07:36刘思邑杨玉姣赵秀丽
基础医学与临床 2024年3期
刘思邑,杨玉姣,杨 涛,赵秀丽
中国医学科学院基础医学研究所 北京协和医学院基础学院 医学遗传学系疑难重症及罕见病全国重点实验室,北京 100005
结节性硬化症(tuberous sclerosis complex,TSC;OMIM 191100或613254)是一种多系统发作的常染色体显性遗传病,发病率为1×10-4~6×10-3,无种族或性别差异[1]。据估计,全世界有约100万人受到该疾病的影响[2]。TSC的临床特征是发生在脑、皮肤、心脏、肾脏和肺等多个器官的错构瘤,以及与之伴随的癫痫、学习困难、行为问题、肾衰竭等并发症状。TSC的临床表现在个体之间可能存在较大差异。几乎所有的TSC患者的病症都会在皮肤上有所体现[3],大约90%的TSC患者有局灶性色素减退,75%的患者有面部血管纤维瘤,50%的患者会出现皮肤增厚的症状,20%~80%的患者指甲会异常生长。超过90%的TSC患者中枢神经系统受到影响,存在癫痫、学习困难、行为问题和自闭症等;60%~80%的TSC患者患有血管平滑肌脂肪瘤、囊肿和肾细胞癌等;30%~50%的TSC患者患有多发性视网膜错构瘤,以及发生在肺部的淋巴管平滑肌瘤病(lymph angioleio myomatosis,LAM)和心脏横纹肌瘤等。此外,部分TSC患者还有其它临床表现,如神经内分泌肿瘤、肝血管平滑肌脂肪瘤和甲状腺乳头状腺瘤,但发生率相对较低。
尽管1910年[4]就有国外学者报道结节硬化症为一种遗传性疾病,但直到20世纪90年代才通过多重连锁分析确定结节硬化症的致病基因为TSC1和TSC2。在所有发现TSC1或TSC2致病变异的TSC患者中,约有三分之二为TSC2变异。另有10%~15%临床诊断为TSC的患者在TSC1或TSC2中均未发现致病变异[5],提示可能存在位于这两个基因非编码区致病变异、或其它新基因的致病变异。
本研究对5个结节性硬化症患者家系进行了TSC1/TSC2致病变异检测,为明确临床诊断、探究遗传病因、以及风险家庭再次妊娠产前基因诊断提供依据。
1 材料与方法
1.1 材料
1.1.1 研究对象:选取2020年1月至2021年7月间在中国医学科学院基础医学研究所进行远程遗传咨询的5个无关TSC家系的8例患者为研究对象。8例患者均表现出不同程度的TSC临床症状,被拟诊为TSC;在获得研究对象(或监护人)知情同意后,收集患者临床资料并采集先证者及其家系成员外周血样本进行致病变异鉴定。本研究获中国医学科学院基础医学研究所伦理委员会批准(编号:015-2015),所有参与者均签署知情同意书。
1.1.2 主要试剂:Tris平衡酚、乙二胺四乙酸二钠、三氯甲烷、琼脂糖、无水乙醇和异丙醇(国药集团化学试剂有限公司);蛋白酶K、dNTP(Sigma-Aldrich 公司);琼脂糖凝胶DNA片段回收试剂盒(Thermo Fisher Scientific 公司);TaKaRa LA Taq with GC Buffer、TaKaRa Ex Taq、TaKaRa DNA Fragment Purification Kit(宝日医生物技术有限公司);PCR引物(北京诺赛基因组有限公司);Nimblegen SeqCap EZ Choice Library试剂盒(Roche 公司)。
1.2 方法
1.2.1 提取基因组DNA:采集先证者及其家系成员外周血(3 mL/人份),EDTA抗凝,采用常规酚-氯仿法提取基因组DNA,经Nanodrop进行纯度和浓度鉴定,-20 ℃保存备用。
1.2.2 基因组panel测序检测候选致病变异:采用包括TSC1/TSC2等154个本实验室重点关注单基因病候选致病基因的个性化定制基因组测序panel进行目标区域捕获,对先证者进行候选基因靶向测序(panel sequencing,PS)。使用1%的琼脂糖电泳检测DNA样品的纯度及完整性,经Qubit®3.0 Flurometer(Life Technologies)检测DNA样品浓度。取200 ng检测合格的DNA模板,根据Nimblegen SeqCap EZ Choice Library方法及流程进行Illumina文库制备,使用 Agilent 2100对文库的插入片段长度进行检测,片段合格后再对文库进行Q-PCR准确定量。在安诺优达基因科技有限公司Illumina HiSeq X Ten平台进行候选基因靶向测序,通过FastQC工具对测序数据进行质控。使用Burrows-Wheeler Aligner(BWA)软件将测序得到的序列与人类基因组参考序列hg19/GRCh37比对;过滤千人基因组(1000G; http://browser.1000genomes.org)、ExAC(http://exac.broadinstitute.org/)及gnomAD(http://www.gnomad-sg.org/)等多个人群数据库和测序公司的本地数据库的高频变异,再经SIFT(http://provean.jcvi.org/seq_submit.php)、Polyphen2(http://genetics.bwh.harvard.edu/pph2/)、CADD(http://cadd.gs.washington.edu/)等软件进行致病性预测,通过ClinVar(https://www.ncbi.nlm.nih. gov/clinvar/)、OMIM(https://omim.org)和 HGMD(http://www.hgmd.cf.ac.uk/ac/index.php)等疾病相关数据库及文献检索变异是否被收录,结合患者临床表型、疾病遗传模式、变异位点致病性等信息筛选候选变异位点。
1.2.3 候选致病变异的验证
1.2.3.1 聚合酶链式反应(PCR)引物设计:在UCSC网站(http://genome.ucsc.edu/)获得TSC1(NM_000368.4)和TSC2(NM_000548.3)的参考序列,应用Primer Primier3(http://primer3.ut.ee/)针对PS测序发现的候选致病变异设计PCR引物(表1),扩增区域涵盖候选变异位点所在的外显子及侧翼序列。
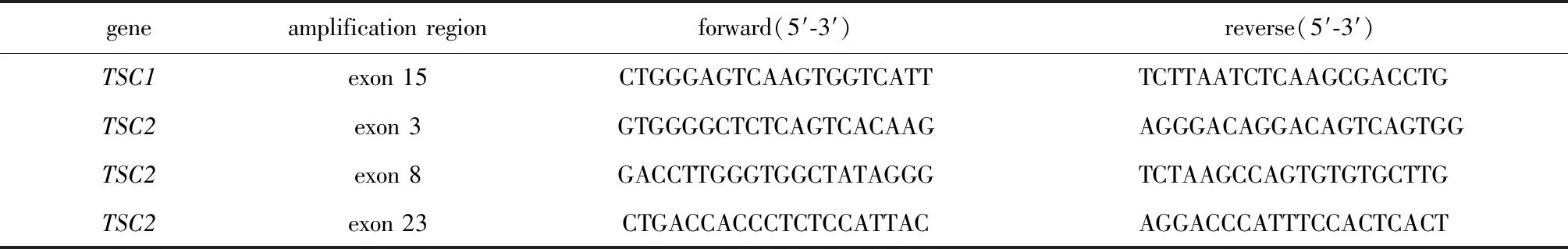
表1 候选致病变异验证PCR引物Table 1 PCR primers for validation of the candidate pathogenic variants
1.2.3.2 PCR反应及测序分析:通过PCR扩增先证者及家系成员候选变异所在的基因区域。PCR反应体系为25 μL,包含50 ng的基因组DNA,12.5 μL的2×GC buffer Ⅰ,4 μL的2.5 mmol/L的dNTP,上下游引物各1 μL(10 μmol/μL),0.25 μL LA Taq(5 U/μL)。扩增条件:95 ℃预变性3 min;35个循环:94 ℃变性30 s,58 ℃退火30 s,72 ℃延伸50 s;72 ℃延伸7 min。PCR产物送往诺赛基因组研究中心进行Sanger测序。测序结果经CodonCode Alingner软件与参考序列比对以检出变异。
1.2.4 变异致病性的分析:参考HGMD数据库和相关文献的研究结果,并依据美国医学遗传学与基因组学学会(The American College of Medical Genetics and Genomics,ACMG)指南对候选变异位点致病性进行判定。在HGMD中查询检出的变异,若变异已被HGMD收录,则依据相关文献所提供的致病性证据结合ACMG标准进行致病性再判定;若未被HGMD收录,则依据ACMG指南所定义的证据链合并分析,如根据VarCards等分析软件预测候选变异的致病性[6]、变异在千人基因组、ExAC及gnomAD等多个人群数据库中的频率、以及变异在家系中与疾病共分离情况等多个分级证据的合并分析结果,确定变异的致病性。
2 结果
2.1 患者临床表现
来自5个家系的8例TSC患者均表现局灶性色素减退。2例先证者有家族史,其余3例为散发病例。5位先证者已在其就诊医院进行多项相关临床检查,结果显示不同程度的多系统TSC特征病变(表2)。家系1先证者有色素脱失斑、癫痫、心脏功能异常,面部有血管纤维瘤(图1A, B);家系2先证者患有癫痫和身体运动障碍,但其母亲只有局部皮肤脱色斑和面部血管纤维瘤;家系3先证者有色素脱失斑、肾脏、中枢神经系统、心脏功能3方面异常;家系4先证者在多个器官,包括大脑、皮肤、心脏、肝脏、肾脏、肺和牙龈中均探测到错构瘤(图1C, D),并有严重的神经症状如癫痫、学习困难、行为等问题;家系5的先证者仅表现局部色素减退,CT检测出其颅内结节钙化但无神经层面表现。其父亲及弟弟亦表现局部色素减退,但未作进一步临床检查。
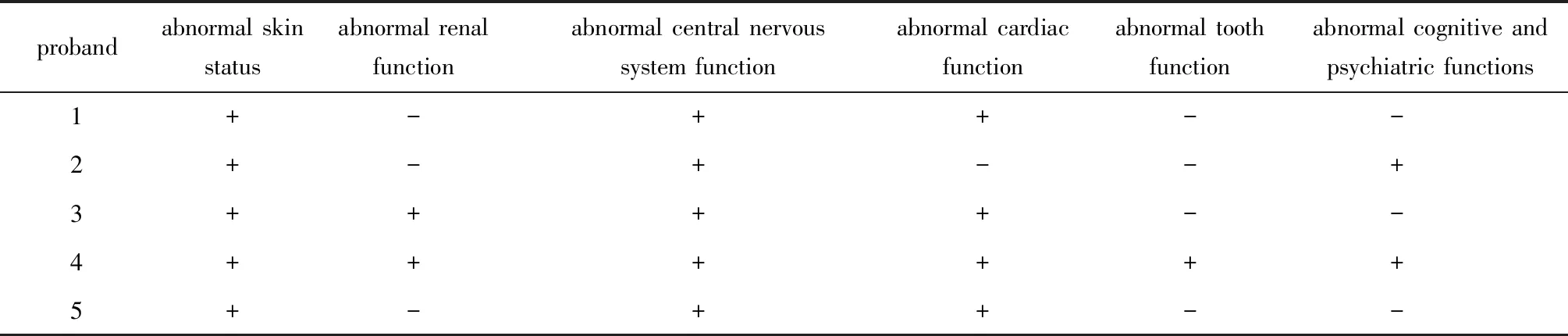
表2 TSC先证者临床表现Table 2 Clinical manifestation of the TSC probands
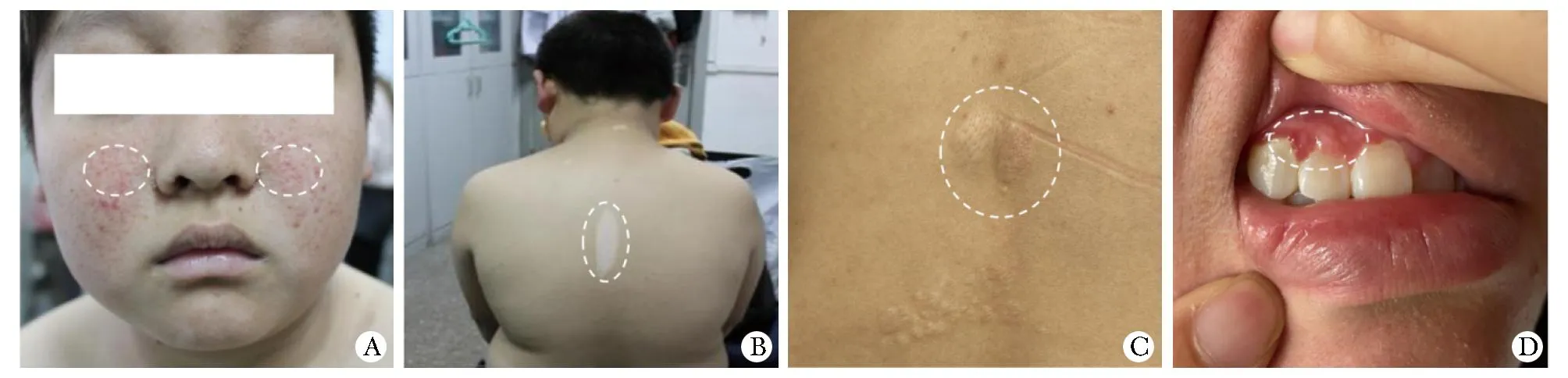
A.facial angiofibroma (family 1 proband, Ⅱ-1); B.depigmentation spots on skin (family 1 proband, Ⅱ-1); C.shagreen patch (family 4 proband, Ⅱ-1); D.odontogenic fibroma (family 4 proband, Ⅱ-1).
2.2 鉴定致病变异
在5个TSC家系中检出致病变异,包括1个TSC1变异和4个TSC2变异(表3)。其中2个为新变异,包括一个无义变异TSC2:c.245G>A(Trp82*)(家系1)和一个移码变异TSC2:c.235delG(Val79Lysfs27*)(家系2)(图2,3)。其余3个为已报道致病变异,已收录于HGMD数据库,包括1个错义变异TSC2:c.2 713C>T(Arg905Trp)(家系3)、一个移码变异TSC2:c.826_827delAT(Met276Valfs60*)(家系4)和一个无义变异TSC1:c.2 074C>T(Arg692*)(家系5)。来自家系1、3和4的变异为新生变异。家系2和家系5为家族遗传病例,变异和疾病呈现共分离。根据ACMG指南并考虑文献提供的致病性判定依据进一步确认了3个已报道变异的致病性(表3)。2个新变异,c.245G>A(p.Trp82*)和c.235delG(p.Val79Lysfs27*),均可产生提前终止密码子而导致所编码蛋白质仅保留了N端不超过全长1/10长度的片段,突变点后多个重要的功能域缺失,丧失其正常功能(非常强致病性证据1,PVS1);在1 000G、ExAC及gnomAD等多个人群数据库中未收录(中等强度致病性证据2,PM2);文献报道Trp82氨基酸位置其它核苷酸变异所导致的无义变异致病(强致病性证据1,PS1)[7]。合并上述分级证据判定这2个新变异为致病变异。
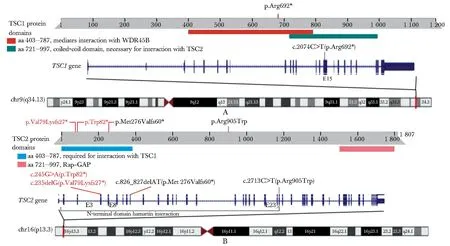
A.pathologic variant detected in TSC1; B.pathologic variants detected in TSC2.

表3 5个TSC家系变异致病性判定Table 3 Pathogenicity determination of the variants detected in 5 TSC families
3 讨论
TSC1位于染色体的9q34,由21个外显子组成,编码Hamartin蛋白;TSC2定位于染色体16p13.3,包含41个外显子,编码Tuberin蛋白(图3)。Hamartin和Tuberin通过它们位于N端区域附近的结构域相互作用[12],并与TBC1结构域家族成员7(TBC1D7)一起形成异三聚体复合物(又称TSC蛋白复合物),是mTORC1信号的主要负调控因子。而Rheb作为小Ras相关的GTP酶, 在活性状态下会激活mTORC1。Hamartin-tuberin复合物可以作为GAP(GTP酶激活蛋白)通过灭活Rheb来抑制mTOR通路[3]。TSC1或TSC2的致病变异破坏了其蛋白产物形成的功能复合物,导致了mTOR通路的过度激活、细胞周期调控异常和错构瘤的形成。
文献报道TSC2致病变异有比TSC1更高的频率和更重的症状[3]。本研究结果与之类似,5个家系中4个为TSC2变异,且患者的表型均比TSC1变异的患者更为严重。由于TSC2编码的Tuberin蛋白(1 807 AA)远远长于TSC1编码的Hamartin蛋白(1 164 AA),也大大增加了TSC2突变的概率,无论是新生变异还是人群中累积的变异都以TSC2居多,故造成TSC2突变患者比TSC1突变患者有更高的比例。TSC1和TSC2编码的Hamartin-Tuberin复合物通过Tuberin蛋白的GAP相关结构域催化Rheb上携带的GTP水解为GDP,从而使得Rheb失活而不能激活mTORC1的激酶活性。因此TSC2变异可能比TSC1变异对Hamartin-tuberin复合物的GAP活性具有更大的破坏性,导致mTOR途径的更大失调[13-14]。本研究中发现的2个TSC2新变异:c.245G>A(Trp82*)和c.235delG(Val79Lysfs27*),可导致蛋白产物缺失下游约9/10的序列,其中包括Rap-GAP及其他重要的结构域(图3)。由于与TSC1互作的区域存在于TSC2上游,有可能这2种缺乏正常功能的TSC2突变蛋白仍然能够结合TSC1,从而导致突变Hamartin-Tuberin复合物缺乏正常功能并干扰了杂合子中正常Hamartin-Tuberin复合物的形成,但确切的致病机制需要更多的功能实验来研究。
TSC1和TSC2的大多数变异由单碱基对缺失或插入和无义变异组成,导致蛋白质合成的过早终止或无义变异介导的mRNA降解。本研究中仅家系3为错义突变TSC2:c.2 713C>T p.(Arg905Trp) ,多篇文献已报道其为致病变异;此外,有研究者[15]通过对野生型和突变型TSC2蛋白的对比研究,发现该突变降低了Tuberin抑制S6K连接域磷酸化的能力。本研究中患者为新生突变,父母表型正常,进一步为该变异的致病性提供了证据。
本研究在5个TSC家系中检出致病变异,包括2个TSC2新变异和3个已报道的致病变异,可为相关家庭的遗传咨询和产前诊断提供依据; 2个新变异的发现丰富了TSC2致病变异谱,有助于提高结节硬化症的诊断率,并可能为疾病的精准治疗提供新的靶点。
猜你喜欢
临床输血与检验(2022年3期)2022-06-22
郑州大学学报(医学版)(2019年3期)2019-06-03
传染病信息(2019年2期)2019-05-17
湖南畜牧兽医(2016年3期)2016-06-05
兽医导刊(2016年12期)2016-05-17
广东海洋大学学报(2015年4期)2016-01-13
听力学及言语疾病杂志(2015年5期)2015-12-24
首都医科大学学报(2015年4期)2015-12-16
重庆医学(2015年12期)2015-03-05
郑州大学学报(医学版)(2015年2期)2015-02-27
