
综合生物信息学分析揭示乳腺癌细胞衰老的预后及免疫特征
2024-03-20 09:41王科江李雪森西南医科大学基础医学院肿瘤医学研究所泸州646000
中国免疫学杂志 2024年2期
王科江 李雪森 (西南医科大学基础医学院肿瘤医学研究所,泸州 646000)
乳腺癌是全球范围内高度流行的癌症。过去二十年,早期诊断工具和治疗方法进步使乳腺癌病死率与1990年相比降低了1/3[1-2]。但乳腺癌仍是女性患者癌症死亡的主要原因,威胁全球妇女的健康。越来越多的证据表明乳腺癌具有高度异质性,局部肿瘤微环境对肿瘤发展至关重要,包括乳腺癌起始、进展、转移及耐药[3]。
细胞衰老是衰老的关键过程之一,是衰老与癌症间的纽带[4]。衰老与肿瘤的联系复杂,目前知之甚少。细胞衰老在肿瘤发生和发展过程中具有“双刃剑”作用。一方面,衰老细胞进入永久性细胞周期停滞的背景下,衰老确保组织动态平衡并防止肿瘤发生[5-6]。另一方面,衰老细胞通过促进多种类型免疫抑制细胞积累及各种炎症相关信号分子和细胞因子激活改变肿瘤微环境,广泛影响肿瘤微环境和肿瘤生长[7-8]。值得注意的是,衰老和细胞衰老可改变免疫细胞健康状况,并最终影响癌症治疗效果,尤其是免疫治疗。但细胞衰老与肿瘤微环境的关系尚不清楚,细胞衰老相关基因在评价乳腺癌免疫浸润和临床预后方面的价值有待进一步研究。
为系统评估乳腺癌患者细胞衰老与预后的相关性,本研究建立了基于细胞衰老相关基因的新风险模型,并探讨了其作为预测预后生物标志物的潜在重要性,基于细胞衰老相关特征深入分析了风险亚群、免疫检查点和免疫细胞渗透间的关系,为乳腺癌免疫治疗策略及细胞衰老调控机制提供了新的见解。
1 资料与方法
1.1 数据来源与下载 从TCGA网站(https://portal.gdc.cancer.gov/)下载乳腺癌RNA-seq转录组数据和相关临床信息,对mRNA count数据进行标准化,将标准化的count数据转化为TPM,进一步对TPM数据进行log2对数转化用于后续分析。将表达数据与临床数据相匹配,去除重复样本,共113例正常样本和1 050例肿瘤样本纳入分析(表1),从GEO数据库(https://www.ncbi.nlm.nih.gov/geo/)下载并整理具有预后信息的327例乳腺癌患者队列(GSE20685)作为外部验证集。细胞衰老相关基因来自CellAge数据库(https://genomics.senescence.info/cells/),该数据库收录了278个经实验验证的细胞衰老相关基因。

表1 TCGA-BRCA队列临床信息基线资料Tab.1 Baseline clinical information of TCGA-BRCA cohort
1.2 差异显著的细胞衰老相关基因鉴定及富集分析 R软件“DEseq2”包鉴别差异表达的细胞衰老相关基因[9],cut-off值设为|log2FC|>1,Padj<0.05,利用R软件“clusterProfiler”包对差异表达的细胞衰老相关基因进行GO和KEGG通路富集分析[10]。
1.3 细胞衰老相关基因预后风险模型构建及外部数据集验证 TCGA中1 050个乳腺癌患者样本作为训练集,采用单因素Cox回归分析评估TCGA中差异表达的细胞衰老相关基因与患者总生存期(OS)的关系,筛选P<0.05的所有基因进行进一步分析。采用LASSO分析选择OS相关的最佳预后基因。通过多因素Cox回归分析得到最优模型,计算风险评分,风险评分=基因表达量1×Coef1+基因表达量2×Coef2+……+基因表达量n×Coefn(Coef:基因在多因素Cox回归分析中的回归系数,n:预后相关细胞衰老相关基因总数)。根据风险评分中位值将患者分为高、低风险组。将GSE20685的327个乳腺癌患者队列作为外部验证集,将基因表达数据带入TCGA训练集风险评分公式计算各样本风险评分进行验证。利用“survival”“survminer”“pheatmap”包绘制TCGA训练集和GSE20685外部验证集风险评分曲线图、风险评分散点图、生存曲线图、高低风险评分组细胞衰老相关基因表达热图。
1.4 预后模型性能评估 在TCGA队列中对风险评分和相关临床参数进行单因素和多因素Cox回归分析,确定风险评分和相关临床参数是否可作为乳腺癌患者OS的独立预测因子。纳入临床病理参数和风险评分利用R软件“regplot”包生成预后列线图预测TCGA中乳腺癌患者一年、三年和五年OS,并采用校准曲线验证预测的生存概率与实际观察结果的吻合度。采用ROC曲线评价风险评分及相关临床参数预测率。
1.5 GSEA基因集富集分析 GSEA可通过将预定义的基因集与特定表型进行比对找出预定义基因集相关表型。根据细胞衰老关键预后基因风险评分中位值进行分组,并与C2.CP.KEGG.v7.2基因集、Hallmark Gene Sets(癌症特征基因集合)基因集进行比对,C2.CP.KEGG.v7.2基因集和Hallmark收集自分子签名数据库(MSigDB),利用R软件“clusterProfiler”包进行分析[10],探索细胞衰老关键预后基因在乳腺癌中可能的作用机制。显著富集的基因集符合以下标准:P<0.05,FDRq<0.25。
1.6 免疫微环境及免疫细胞浸润分析 利用“CIBERSORT”包反卷积算法评估所有TCGA乳腺癌病例中免疫细胞浸润比例,并绘制免疫细胞浸润比例丰度图和22种免疫细胞相关性热图[11]。根据细胞衰老关键预后基因风险评分中位值进行分组,评估22种免疫细胞在高、低风险组的浸润比例和免疫检查点基因表达。利用“ESTIMATAE”包进行免疫微环境分析,计算免疫评分,评估风险评分与免疫微环境的关系[12]。
1.7 体细胞突变分析 体细胞突变数据来源于TCGA(https://portal.gdc.cancer.gov/),数据类型为“Masked_Somatic_Mutation”,利用“maftools”包分析TCGA体细胞突变数据[13],计算基因突变频率。
1.8 统计学分析 所有统计分析和数据绘图均采用R软件(4.1.2)进行。
2 结果
2.1 TCGA数据库中差异表达的细胞衰老相关基因鉴定及差异表达基因功能富集分析 278个细胞衰老相关基因中有68个基因在肿瘤组织和正常样本中的表达存在显著差异,其中36个基因表达上调,32个基因表达下调(图1A、B)。通过功能富集分析探讨上述68个细胞衰老差异表达基因在TCGA-BRCA队列中的生物学功能和显著富集通路,GO及KEGG富集分析结果显示,差异基因显著富集于细胞周期、细胞衰老相关信号通路及生物学进程(图1C、D)。
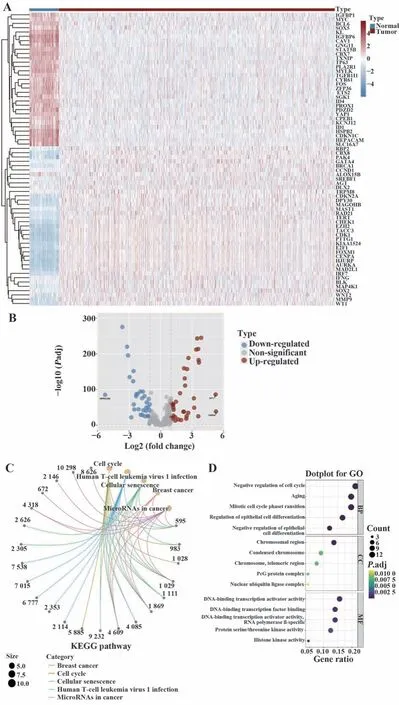
图1 细胞衰老相关基因差异分析及GO/KEGG富集分析Fig.1 Difference analysis of senescence related genes and GO/KEGG enrichment analysis
2.2 细胞衰老相关基因预后模型构建及验证 在TCGA队列中对上述68个细胞衰老差异基因进行单因素Cox回归分析,结果显示8个基因与乳腺癌患者OS相关(P<0.05,图2A)。为避免过度拟合,对8个有预后价值的基因进行LASSO回归(图2B、C),对LASSO回归确定的7个预后相关基因进行多因素Cox回归,确定了WT1、IFNG、TP63、IGFBP6、CPEB1 5个细胞衰老关键预后基因的最佳预后模型,风险评分=WT1×0.162 094+IFGN×(-0.320 910)+TP63×(-0.120 800)+IGFBP6×(-0.141 510)+CPEB1×0.385 455,预后模型森林图如图2D所示。根据中位风险评分将患者分为高、低风险组,风险评分散点图、风险评分曲线图显示随着风险评分升高,相同时间点患者死亡率越高(图3A、B),热图展示了5个基因在高、低风险组的表达(图3C),生存曲线显示高风险组预后较差(图3D)。为验证预后模型,对来自GEO数据库的GSE20685乳腺癌队列按照与TCGA队列相同的风险评分计算公式计算各样本风险评分,根据TCGA队列中位评分将GSE20685验证队列分为高、低风险组,结果显示构建的模型具有普适性,在GSE20685队列中得到了验证(图3E~H)。
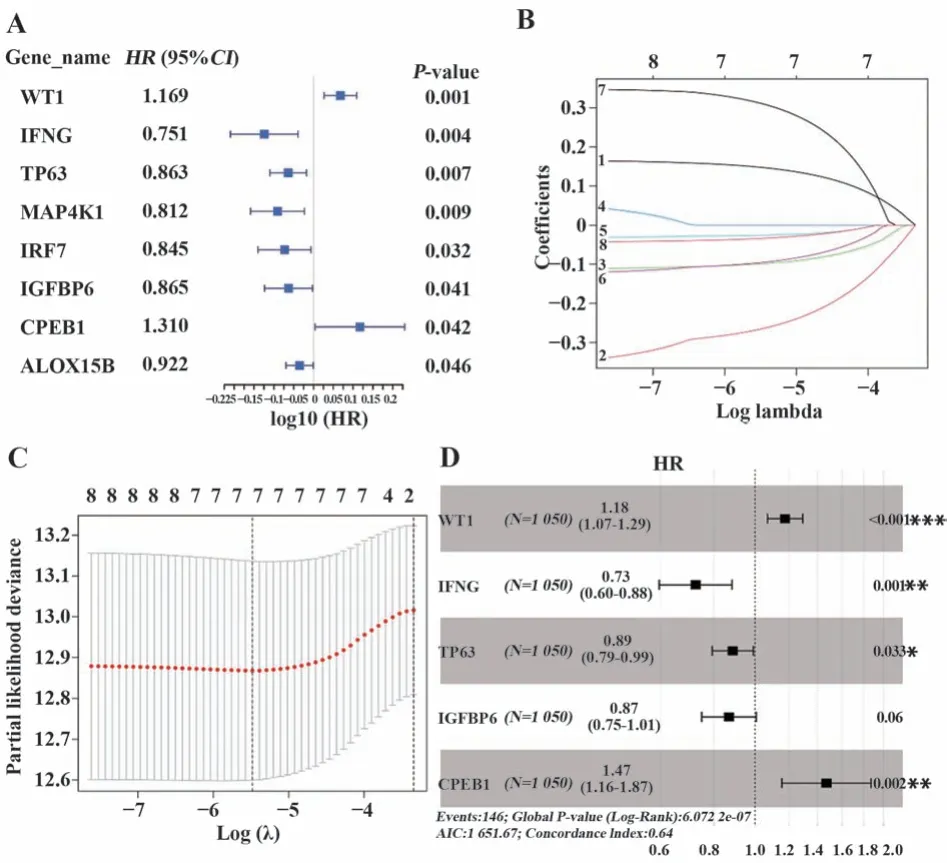
图2 细胞衰老相关基因预后风险模型构建Fig.2 Construction of a prognostic risk model for cell senescence related genes
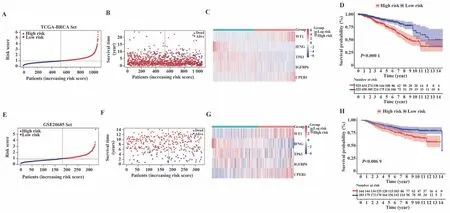
图3 TCGA训练集和GSE20685外部验证集高、低风险评分组患者基因表达及生存状态评估Fig.3 Evaluation of gene expression and survival status of patients in TCGA training set and GSE20685 external validation set with high and low risk score
2.3 预后模型性能评估及列线图建立 为进一步验证预后模型的准确性,研究风险评分是否可作为TCGA-BRCA队列的OS独立预后因素,对临床特征和风险评分进行单因素Cox回归分析和多因素Cox回归分析,结果显示仅风险评分、年龄、淋巴结转移分期(N_stage)在单因素Cox回归分析和多因素回归分析中均为独立预后因素(P<0.001,图4A、B)。纳入风险评分、年龄、淋巴结转移分期3个独立预后因子构建预后列线图(图4C),校准曲线提示该列线图预测生存率与队列实际生存率拟合度较好(图4D)。绘制多重ROC曲线比较风险评分和临床参数预测患者预后准确率,结果显示,风险评分具有较好的预测能力(图4E)。
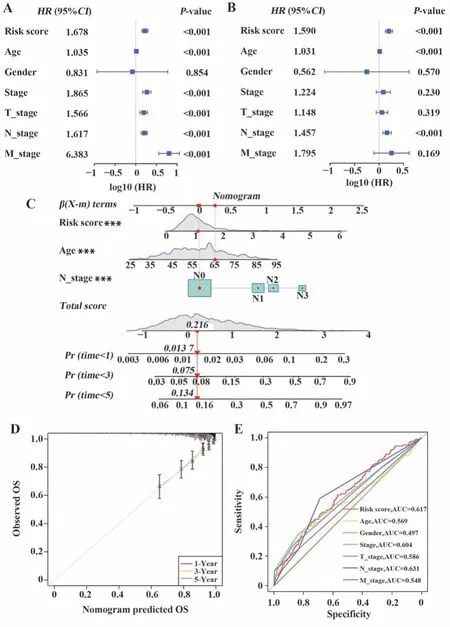
图4 预后风险评分模型的独立预后价值Fig.4 Independent prognostic value of prognostic risk score model
2.4 GSEA富集分析 为进一步探讨风险模型分类亚组间基因功能和通路差异,根据风险评分中位值将患者分为高、低风险组进行GSEA富集分析,选择显著富集的前10个基因集进行展示,结果显示肿瘤免疫相关特征基因集和信号通路在高风险组中显著下调,如KEGG数据集中的抗原加工与呈递、趋化因子信号通路、细胞因子及细胞因子受体相互作用(图5A),Hallmark基因集(癌症特征基因集合)中的IL2_STAT5信号通路、IL6_JAK_STAT3信号通路,提示高风险患者可能处于免疫抑制状态(图5B)。
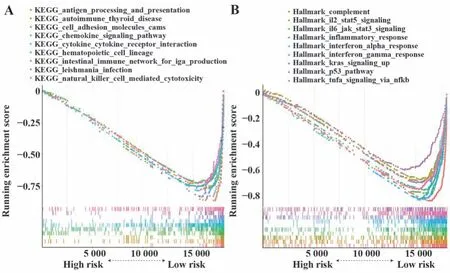
图5 风险得分相关基因显著富集于肿瘤免疫相关通路Fig.5 Genes related to risk score were significantly enriched in tumor immune-related pathways
2.5 高、低风险患者免疫微环境分析 基于GSEA分析结果推测高、低风险患者肿瘤免疫微环境可能存在差异。应用CIBERSORT反卷积算法进一步确认风险评分与免疫成分的关系,构建乳腺癌患者22种免疫细胞丰度图(图6A),分析22种免疫细胞的相关性(图6B)。进一步分析高风险患者和低风险患者22种免疫细胞浸润比例,结果显示CD8 T细胞、M0巨噬细胞、M1巨噬细胞、M2巨噬细胞比例存在显著差异(P<0.001,图6C)。产生活性氧的M1巨噬细胞是肿瘤生长抑制因子,而产生IL-10和TGF-β的M2巨噬细胞可促进肿瘤生长[14]。许多肿瘤中,如结直肠癌、乳腺癌、卵巢癌或膀胱癌,CD8 T细胞浸润是很好的预后标志[15]。值得注意的是,本研究中高风险患者M2巨噬细胞显著多于低风险患者,CD8 T细胞、M1巨噬细胞显著少于低风险患者(P<0.001,图6C),表明基于风险评分对乳腺癌患者进行风险分层得到的结果与既往研究相符,风险评分可作为提示肿瘤微环境免疫细胞浸润和预测患者预后的指标。为评估高、低风险患者对免疫治疗的影响,探讨风险评分与公认的免疫检查点基因的相关性,结果显示低风险患者PDCD1、CD274、CTLA4、CD86、LAG3、HAVCR2、TIGIT表达高于高风险患者(图6D);提示低风险患者免疫检查点基因表达高于高风险患者,低风险患者可能有更好的免疫治疗反应。进一步利用R软件包ESTIMATE根据基因表达计算各肿瘤中各患者免疫评分,比较高、低风险组免疫评分差异及风险评分与免疫评分的相关性,结果显示高风险组免疫评分显著低于低风险患者,且风险评分与免疫评分呈负相关(图6E、F)。提示高风险患者肿瘤微环境倾向于免疫抑制状态,随着风险评分升高免疫评分降低,同时高风险患者伴随促肿瘤生长的肿瘤相关巨噬细胞高度浸润及免疫检查点基因表达显著降低,可能影响高风险患者免疫治疗应答,尤其是免疫检查点抑制剂。

图6 高、低风险患者免疫微环境分析Fig.6 Analysis of immune microenvironment in patients with high and low risk
2.6 高、低风险患者基因组突变分析 结果展示了高、低风险亚组前30个高频突变基因,高风险组460个样本中420个样本发生了突变(91.3%),低风险组466个样本中422个样本发生了突变(90.56%),其中高风险患者抑癌基因TP53突变频率(38.00%)高于低风险(31.00%,图7A、B红框部分)。为进一步说明TP53基因在两组队列中的突变差异,使用“Maftool”包的“mafCompare”函数比较两组间TP53差异突变情况,该函数对两个队列生成的2×2关联表进行fisher检验,找出差异突变基因,高风险组中TP53基因173个样本发生了突变,低风险组中143个样本发生了突变,OR<1(OR=0.735)且P<0.05(P=0.027),表明高风险患者抑癌基因TP53突变频率更高。

图7 高低风险亚组基因突变瀑布图Fig.7 Waterfall map high and low risk subgroups
3 讨论
乳腺癌是世界最常见的恶性肿瘤,据最近癌症数据统计,乳腺癌占所有新诊断恶性肿瘤的30%以上,尽管乳腺癌在诊断、手术和药物开发方面取得了一定进展,但治疗反应不充分及复发和转移,乳腺癌治疗仍面临严峻挑战[16]。此外,多数治疗方法均针对癌细胞开发,忽略了肿瘤微环境周围的非癌症成分,导致反应结果较差,肿瘤微环境包括各种非恶性细胞和非细胞成分,包括免疫系统成分、血细胞、内皮细胞、脂肪细胞和间质成分,可能以不同方式改变肿瘤行为[17]。衰老是一个复杂的生物过程,具有细胞自主性和旁分泌效应,对肿瘤微环境有重大影响[7]。但乳腺癌中衰老细胞如何与肿瘤免疫细胞相互作用及其在评估肿瘤免疫浸润性和临床预后方面的价值尚未见报道。
本研究分析了乳腺癌中细胞衰老相关基因的表达模式、预后价值及对肿瘤免疫微环境的影响,使用LASSO-Cox构建了新的基于TCGA数据集中5个衰老特征表达的生存预测模型,该模型在GEO数据库的公开数据集GSE20685得到了很好的验证。值得注意的是,本研究发现单因素及多因素Cox回归提示风险评分是乳腺癌患者的独立预后因素,根据风险评分中位值分组的高、低风险患者免疫状态具有高度异质性,高风险患者伴随促肿瘤生长的肿瘤相关巨噬细胞高度浸润以及免疫检查点基因表达显著降低,具有良好预后提示的CD8 T细胞浸润丰度降低,且高风险患者基于ESITIMATE算法得出的免疫评分显著低于低风险患者。提示细胞衰老相关基因预测模型指导乳腺癌患者预后具有积极作用,可与特异性免疫检查点因子或肿瘤微环境联合作为预测免疫检查点抑制剂反应的生物标志物。
本研究模型中共纳入5个细胞衰老相关基因:WT1、IFNG、TP63、IGFBP6、CPEB1。研究发现,原癌基因KRAS诱导的肿瘤模型中,WT1的RNA干扰显著降低了肿瘤负担,诱导了细胞衰老,并抑制了肿瘤细胞增殖[18]。WT1还通过调节肌动蛋白活性影响细胞骨架重排,参与细胞运动,提示WT1在癌细胞侵袭和迁移中可能发挥作用[19]。IFNG属于Ⅱ型干扰素家族成员,由免疫细胞如T细胞和NK细胞产生,通过激活效应免疫细胞和增强抗原呈递,在抗菌、抗病毒和抗肿瘤反应中发挥关键作用[20]。临床研究发现结直肠癌患者外周血单个核细胞中TNF和IFNG均受到抑制,复发性结直肠癌患者外周血单个核细胞IFNG表达降低[21]。TP63作为TP53、TP63和TP73家族基因成员之一,在转录调控、诱导细胞凋亡、细胞周期停滞、衰老、代谢重编程和干细胞维持方面发挥重要作用[22]。此外,TP63的两个亚型TAp63和DNp63在干细胞维持、肿瘤转移中发挥不同调节作用[23]。研究发现TAp63在13个肿瘤发展特征中始终表现出肿瘤抑制模式,而DNp63在肿瘤发生发展中作为肿瘤抑制基因或癌基因具有双重作用[24]。IGFBP6是一种IGF结合蛋白,可延长IGF半衰期,并可抑制或刺激IGF在细胞培养中的促生长作用,IGFBP6可能在结肠癌发展过程中作为一种抑癌基因,低表达IGFBP6可作为结肠癌的独立预后生物标志物[25]。另一项研究发现对TMZ(替莫唑胺)敏感的胶质瘤细胞分泌的IGFBP6驱动旁分泌机制在体内外抑制表达IGF1-R的TMZ耐药细胞增殖,耐TMZ胶质瘤细胞中IGFBP6过表达延长了生存期[26]。CEBP1是编码胞质多聚腺苷酸化元件结合蛋白家族成员,研究发现CPEB1是肿瘤细胞(Hela)进入M期和细胞增殖所必需的,单独去除其家族成员CPEB4对细胞周期的影响很小,但CPEB4与CPEB1共同耗尽时可观察到对细胞增殖的协同抑制[27]。相反的是,研究发现CPEB1表达下调促进乳腺癌细胞增殖、转移和细胞周期进展,且CPEB1通过介导SIRT1/SOX2通路调控乳腺癌发生[28],但CPEB1也能在肿瘤细胞中发挥促增殖作用,前提是CPEB1首先在原代细胞中诱导细胞衰老[29],提示CPEB1在肿瘤中的调控作用可能与衰老微环境相关。进一步证实细胞衰老相关基因在肿瘤中的调控是双向的,发挥抑癌或促癌作用的关键可能取决于肿瘤微环境差异。
另外,本研究虽提供了一定理论基础和研究参考,但也存在一定局限性。利用公共数据库的回顾性数据对现有模型进行开发和验证,尚需更多前瞻性研究数据验证其临床应用,上述发现还需进一步研究证实。其次,这些细胞衰老关键预后基因在预后模型中调节乳腺癌进程的潜在机制尚不清楚,其生物学功能需进一步探索。最后,由于细胞衰老在癌症中的作用很大程度上被忽视,因此更广泛地了解癌症、衰老和免疫环境间的联系非常重要。本研究确定并验证了基于5个细胞衰老相关基因的细胞衰老相关信号,作为免疫细胞浸润指标对乳腺癌具有独立预后价值。因此,识别影响肿瘤免疫应答的细胞衰老相关基因并进一步研究其调控机制可能有助于危险分层,并为提高乳腺癌对免疫治疗的反应提供靶点。
猜你喜欢
中老年保健(2022年6期)2022-08-19
疯狂英语·新读写(2021年10期)2021-12-07
小学生学习指导(低年级)(2020年4期)2020-06-02
软件(2020年3期)2020-04-20
奥秘(2019年8期)2019-08-28
中国生殖健康(2019年2期)2019-08-23
中国生殖健康(2019年6期)2019-01-06
军营文化天地(2018年2期)2018-12-15
祝您健康(2018年5期)2018-05-16
产品可靠性报告(2017年7期)2017-09-05
