
A rare missense PAX6 mutation causes atypical aniridia in a three-generation Chinese family
2024-03-20 06:33ZhiBoLinChunYunFengJinLiAnPengPanHaiSenSunYongYuShiHaoChen
Zhi-Bo Lin, Chun-Yun Feng, Jin Li, An-Peng Pan, Hai-Sen Sun, A-Yong Yu, Shi-Hao Chen
1National Clinical Research Center for Ocular Diseases, Eye Hospital, Wenzhou Medical University, Wenzhou 325027,Zhejiang Province, China
2Department of Ophthalmology, the Quzhou Affiliated Hospital of Wenzhou Medical University, Quzhou People’s Hospital,Quzhou 324000, Zhejiang Province, China
Abstract
● KEYWORDS: PAX6 gene; atypical aniridia; missense mutation; mutation
INTRODUCTION
Congenital aniridia is a rare genetic eye disorder that affects up to 1 in 64 000 individuals worldwide and demonstrates no predilection for race or sex.Approximately two-thirds of diagnosed patients are autosomal dominantly inherited and the remaining one-third of cases arise sporadically[1-2].Classical aniridia is characterized by partial or complete bilateral absence of the iris, however other manifestations may also occur, including developmental defects of the cornea, lens, retina, anterior chamber angle, and optic nerve in both eyes.The predominant clinical features include missing irises, nystagmus, foveal hypoplasia, cataracts,glaucoma, aniridia-related keratopathy (ARK), and severely compromised vision[2-4].Moreover, aniridia can manifest as an isolated ocular abnormality or be a part of a systemic disorder such as Wilms tumour-aniridia syndrome, which consists of Wilms’ tumor, aniridia, genitourinary abnormalities, and intellectual disability; Wilms tumour-aniridia syndrome occurs with or without obesity[5-6].
The majority of clinical aniridia cases result from mutations in the paired box gene-6 (PAX6, OMIM: 607108) and its associated regulatory regions[1,3-5,7-10].ThePAX6gene, located on chromosome 11p13, is a member of the highly conserved paired box gene family.It encodes a transcriptional regulator including two DNA binding domains—the paired domain(PD) and the homeodomain (HD)[2,8,10-11]—and is critical for eye, brain, spinal cord, and pancreas development[11-13].So far,more than 500 variants ofPAX6related to aniridia have been identified[1].The majority of these variants involve frameshift, nonsense, or splice site mutations that lead to loss of function nucleotide substitutions or the production of truncated proteins[14-15].PAX6 haploinsufficiency tends to cause typical aniridia, which is characterized by partial or complete iris hypoplasia, nystagmus and foveal hypoplasia, later-onset cataracts, glaucoma, and ARK[2,4,10,16-17].
Many studies have reported that clinical phenotypes of aniridia are highly variable among individuals with both different and identical genotypes[3,9-10,18-20].Because of this high phenotypic variability, it is challenging to establish genotype-phenotype correlations.Moreover, non-PAX6mutations that give rise to aniridia-like phenotypes, such as iris defects with anterior segment dysgenesis, have also been reported[21].Diagnosis of aniridia is straight forward when the typical aniridia phenotype is present, but mild or atypical aniridia phenotypes may hinder clinical diagnosis and subsequent management.Thus, it is important to investigate the underlying mutations present in atypical aniridia in order to improve clinical outcomes for these patients.To investigate the relationship between a particular type ofPAX6mutation and aniridic eye phenotypes,we recruited Chinese family members across three-generations with atypical aniridia.Targeted next-generation sequencing(NGS) and Sanger sequencing were performed to identify the mutation inPAX6.A rare missense heterozygous mutation c.622C>T (p.Arg208Trp) inPAX6was identified in all affected individuals, but not in any of the unaffected family members.
SUBJECTS AND METHODS
Ethical ApprovalOur study was approved by the Ethics Committee of The Eye Hospital of Wenzhou Medical University and was in accordance with the tenets of the Declaration of Helsinki (2021-239-K-209).Written informed consent was obtained from all participants.
Subjects and DNA SpecimensA total of eleven members from a Chinese family, spanning three consecutive generations,with atypical aniridia were recruited from The Eye Hospital of Wenzhou Medical University.This family included eight affected and three unaffected members.Totally 180 unrelated subjects without any eye disease or other known syndromes were recruited as healthy controls.All affected family members underwent a complete ophthalmologic examination that included a best-corrected visual acuity (BCVA) test, slit-lamp examination, intraocular pressure (IOP) measurement, optical coherence tomography (OCT), and fundus examination.Other non-affected family members underwent a simple slit lamp examination.
Peripheral blood samples were collected from the proband and her family members, and genomic DNA was extracted using a DNA Extraction kit (Simgen, Hangzhou, China) following the manufacturer’s protocols.Blood samples were also collected from the healthy controls.
Whole-exome Sequencing and AnalysesDNA samples were subjected to whole-exome sequencing (WES).The genomic DNA was used to generate Illumina libraries in agreement with the manufacturer’s sample preparation protocol.After Novaseq6000 platform (Illumina, San Diego, USA) and IDT’s xGen Exome Research Panel V1.0 (Integrated DNA Technologies, San Diego, USA) sequencing, the clean read sequences were aligned to a human reference genome (hg19/RCh37).Subsequently, single nucleotide polymorphisms,insertions, and deletions were identified using the SOAPsnp software and GATK.
Variant Analyses and IdentificationWe evaluated the putative disease-causing variant using assorted software and online databases[22-23], including: Exome Aggregation Consortium (ExAC, http://exac.broadinstitute.org/), 1000 Human Genome Project (ftp://1000genomes.ebi.ac.uk/vol1/ftp), NHLBI Exome Sequencing Project (ESP, http://evs.gs.washington.edu/EVS/), and dbSNP137 (www.ncbi.nlm.nih.gov/SNP/).The pathogenicity prediction of the candidate variant was assessed using SIFT (http://sift.jcvi.org),Polyphen-2 (http://genetics.bwh.harvard.edu/pph2/), Mutation assessor (http://mutationassessor.org/), MutationTaster (http://mutationtaster.org/), and PROVEAN (http:// prove an.jcvi.org/index.php).Multiple sequences were aligned and compared using Clustal Omega (http://www.ebi.ac.uk/Tools/msa/clustalo/).SMART (http://smart.embl-heidelberg.de/) was used to simulate the topological model of the gene’s polypeptide.Direct Sanger sequencing was used to validate the variant, for segregation analysis in the present family, and was performed in the 180 unrelated controls.Additionally, associated wildtype and mutant protein models were predicted by Phyre2(http:// www.sbg.bio.ic.ac.uk/phyre2/ html/page.cgi? id=index)and further visualized using Pymol Molecular Graphics System (Pymol).
RESULTS
Clinical FindingsThe proband (III:1) was a 14-year-old female that complained of bilateral blurred vision since childhood; she had no history of surgery or medical treatment in either eye.Eye examination revealed that her visual acuity(VA) was 20/400 ocular dexter (OD) and 20/1000 oculus sinister (OS), and her BCVA was 20/100 OD and 20/400 OS.Refractive errors were -6.0 D OD and -4.0 D OS, with axial length (AL) values of 26.1 mm OD and 25.6 mm OS.Her IOPs were 15.7 mm Hg OD and 18.1 mm Hg OS.Slit lamp examination of the anterior segments demonstrated obvious corneal nebula in the pupil, though no stromal iris abnormality was present and both pupils were normally reactive to light.Clouding of the lens was observed in both eyes, which demonstrated minimal cataracts (Figure 1A, 1D).Further examination revealed bilateral tessellated fundus accompanied by peripapillary atrophy which appeared as absence of the foveal pit in the left eye and vessels crossing the foveal area in both eyes (Figure 1B, 1E).B-scan echography results were normal in both eyes (Figure 1C, 1F).The proband also presented with dramatic nystagmus which prevented acquisition of clear OCT images.Nevertheless, a mild degree of foveal hypoplasia in the right eye and absence of foveal contour corresponding to at least grade 2 foveal hypoplasia in the left eye were observed (Figure 1G, 1H).All affected individuals exhibited similar phenotypes as the proband,which included low visual acuity, keratopathy, nearly normal irises, mild cataracts, high myopia, high-frequency nystagmus,and foveal hypoplasia.Phenotypic information pertaining to the eight affected patients is presented in Table 1.None of the family members had any systemic abnormalities.Eleven members, eight diagnosed with atypical aniridia, across three generations of a Chinese family were investigated in the present study.A constructed pedigree revealed an inheritance pattern consistent with autosomal dominant inheritance(Figure 2A).

Figure 1 Clinical features of the proband with a PAX6 mutation A, D: The anterior segments of each eye (A: Right eye, D: Lefteye).Both eyes present with corneal nebula, a complete iris, and mild cataracts.B, E: Fundus photographs demonstrate a tessellated fundus accompanied by peripapillary atrophy.C, F: B-scan echography images are within normal limits (C: Right eye, F: Lefteye).G, H: Results of spectral-domain optical coherence suggest bilateral foveal hypoplasia.The red arrow indicates the structure of the optic disc; the fovea appears more developed in the right eye than in the left.
WES and Segregation Analysis Revealed A Mutation inPAX6WES was performed on all affected individuals except patient II:4 due to lack of a blood sample.The autosomal dominant inheritance pattern observed in the pedigree was further confirmed by Sanger sequencing.All affected family members were heterozygous for thePAX6mutation (c.622C>T,p.Arg208Trp), while unaffected members had a normal genotype (Figure 2B).Moreover, this variant was absent in all 180 unrelated controls.The variant c.622C>T converts the hydrophilic amino acid arginine into tryptophan, a hydrophobic amino acid, at position 208.We compared the mutated region across species and found that this variant occurred in a remarkably conserved region (Figure 2C), indicating its importance for protein function.The p.Arg208Trp substitution was predicted to be pathogenic to the protein using SIFT,PolyPhen-2, and Mutation Taster.
In SilicoAnalysisAll affected family members were found to carry a heterozygous mutation within exon 8 of thePAX6gene.Subdomain identification was carried out using SMART,which demonstrated that this mutation was located just two amino acids upstream of the HOX-domain start (Figure 3A).Comparative and structural analyses of p.Arg208Trp were performed using PyMol to predict the potential pathogenic effect on the protein.3D structural modeling revealed that the replacement of an arginine with a tryptophan in thePAX6protein product destroyed the short amino acid motif LRKR,which may responsible for the pathogenicity of this mutation(Figure 3B).

Figure 2 Identification of a PAX6 mutation in a three-generation family with atypical aniridia A: The pedigree shows the unaffected (white)and affected family members (black) and reveals an autosomal dominant mode of inheritance.Circle: Female; Square: Male.B: Sanger sequencing confirmed segregation of the heterozygous missense mutation, c.622C>T (p.Arg208Trp).C: Evolutionary conservation of the arginine residue in PAX6.Alignments of the mutated region across various species reveals that the pathogenic mutation occurred at a highly conserved position.
DISCUSSION
Congenital aniridia is typically characterized by partial or complete absence of the iris.Some features of aniridia, such as nystagmus and foveal hypoplasia, are present at birth.Furthermore, a patient’s visual impairment is not determined by the degree of iris absence, but rather the degree of foveal hypoplasia, as well as the extent of nystagmus, corneal opacity, glaucoma, and cataract[21].In this study, we report a heterozygous mutation inPAX6(c.622C>T, p.Arg208Trp)associated with atypical aniridia identified across three generations of an affected Chinese family.The ophthalmic abnormalities of the patients were relatively similar and included keratopathy, cataracts, nystagmus, and macular fovea hypoplasia (Table 1).Surprisingly, the irises of the affected patients were essentially intact or even normal, which contrasts with the presentation of classic aniridia.Because of this atypical presentation, definitive diagnosis of these family members relied on accurate genetic testing.This identified mutation has been previously identified as familial and pathological by Hansonet al[24], however they did not report the patient’s clinical features, thus little was known about the effects of the mutation.
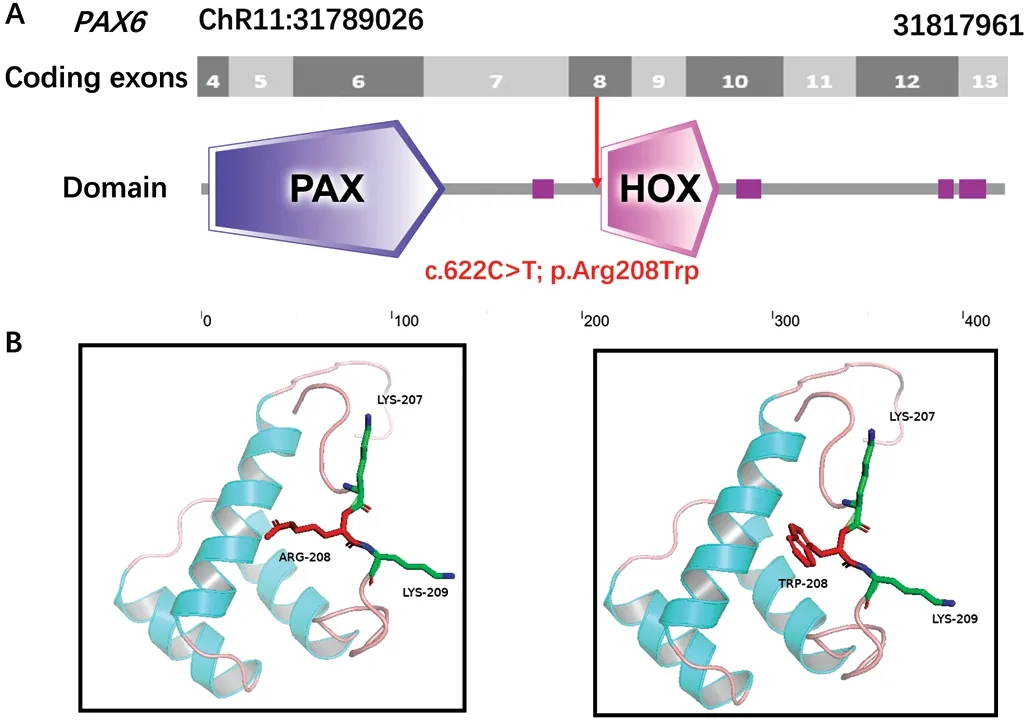
Figure 3 Modeling structure analyses of the identified PAX6 mutation A: Map of the 6944 bp PAX6 gene.Predicted PAX6 domain structure.Pax is indicated in dark blue, Homeobox is indicated in light pink and low complexity region is indicated in dark pink.B: Predicted crystal structure of the wild-type (WT; left) and mutant (right) PAX6 protein.Red represents residue of the WT (left) and mutant (right),while green indicates residues that interact with either PAX6 protein.
PAX6was the first known homeobox gene responsible for aniridia, and it shows extremely high conservation in both coding and non-coding regions across various species[2].In the present study, the arginine at position 208 was also determined to be remarkably conserved as an integral part of the conserved short amino acid motif LRKR (Figure 2C).It has been found that LRKR specifically exists in proteins containing both an intact PD and HD, but is absent from other homeodomain proteins[24].The identified variant p.Arg208Trp alters this conserved structure (Figure 3B).Nonsense or frameshift mutations often result in loss of function due to mutation mediated protein truncation.Certain aniridia phenotypes arise from a missense mutation inPAX6that result in abnormal degradation of the mRNA product[25].Still, the pathogenic mechanism of this mutation remains poorly understood, thus further investigations are needed.PAX6mutant exhibit highly phenotypic variability in both inter- and intra-family.A great deal of research has aimed to elucidate the relationship between genotype and phenotype inPAX6mutations, however definitive correlations remain difficult to establish.It has been previously reported that the most common type of refractive error in aniridia is myopia,and extreme myopia may even be the most prominent aniridia feature[26-27].Overall, particular variants may lead to distinct phenotypes.Loss of function variants—including frameshift,nonsense, and splicing site mutations—ofPAX6are the most common cause of classic aniridia, which is more likely to be associated with severely affected individuals[1,4-5,8-9,28].Milder iris abnormalities are typically attributed to missense mutations[3,17,28-29].Observations of patients with less severely affected irises in this study are thus classified as atypical aniridia.Yokoiet al[30]previously observed no heterogeneity between eyes in the same affected individual, suggesting similar gene regulation in both eyes.This finding is consistent with the ocular characteristics of patients identified in our study.Appropriate patient treatments should be provided depending on their ocular manifestations.For example, iris tissue defects can be repaired with suture or implantation of iris prosthesis according to the amount of iris stump available[31].Treatment of ARK is also determined by its severity; autologous serum and preservative-free lubricants may be useful in early stages,however penetrating keratoplasty is required when ARK affects the visual axis as long as underlying limbal stem cells are still sufficient[21].Cataract is the most common morphological type of lens abnormality, and in these cases, surgery is often helpful.Moreover, medical therapy should be the first choice in patients with complicated glaucoma due to resistance to conventional surgical treatment[32].Future therapeutic directions include gene-targeted therapy for aniridia and other genetic conditions.Regardless, aniridia patients need long term monitoring due to the progressive nature of the disease.

Table 1 Clinical characteristics of the eight related patients with aniridia and one normal family member II:3
Occasionally, cases with strong clinical indication may guide us toward specific molecular investigations ofPAX6.Diagnosis of aniridia was challenging in this family given their roughly normal irises.Thus, molecular diagnosis of familial aniridia is critical, especially in atypical cases or diseases with similar phenotypes which can cause serious complications.Genetic testing is also crucial in sporadic cases, as it identifiesPAX6mutations in approximately 85% of related cases[33].
Our work also has some limitations.High quality OCT examination was not possible due to extreme nystagmus.Additionally, no functional analysis was performed, which could have provided new insights into this variant mechanism.In conclusion, we identified a rare heterozygous (c.622C>T,p.Arg208Trp) variant of thePAX6gene as a putative cause of aniridia and further described its phenotype in this Chinese family.Based on previous reports, our study provides useful clues for confirming the pathogenicity of this mutation and expands understanding of the relationship between a missense mutation ofPAX6and its resulting phenotype.The clinical features of diversePAX6mutations require more attention on account of the development of genetic analysis.
ACKNOWLEDGEMENTS
Conflicts of Interest:Lin ZB,None;Feng CY,None;Li J,None;Pan AP,None;Sun HS,None;Yu AY,None;Chen SH,None.
 International Journal of Ophthalmology2024年3期
International Journal of Ophthalmology2024年3期
- International Journal of Ophthalmology的其它文章
- Meibomian glands segmentation in infrared images with limited annotation
- Artificial intelligence for the detection of glaucoma with SD-OCT images: a systematic review and Meta-analysis
- Overexpression of TRPV1 activates autophagy in human lens epithelial cells under hyperosmotic stress through Ca2+-dependent AMPK/mTOR pathway
- Dry environment on the expression of lacrimal gland S100A9, Anxa1, and Clu in rats via proteomics
- Semaphorin 7A impairs barrier function in cultured human corneal epithelial cells in a manner dependent on nuclear factor-kappa B
- Novel MIP gene mutation causes autosomal-dominant congenital cataract