
单空位缺陷对二维δ-InSe 稳定性的影响*
2024-03-19 00:42苗瑞霞王业飞谢妙春张德栋
物理学报 2024年4期
苗瑞霞 王业飞 谢妙春 张德栋
(西安邮电大学电子工程学院,西安 710121)
二维InSe 半导体材料由于其优异的电学性能以及适中可调的带隙等优点,引起了研究者的关注.材料中的空位缺陷不仅影响材料的光电学特性,还影响材料的环境稳定性.相比于InSe 材料中的其他相,δ-InSe 具有更优异的材料性能,然而关于对该材料环境稳定性影响的研究未见报道.本文基于密度泛函理论,系统研究了O2 环境下二维δ-InSe 材料的稳定性问题.结果表明: 1)在O2 环境下,完美δ-InSe 表面具有良好的惰性和稳定性,O2 分子在其表面从物理吸附到解离吸附需要克服1.827 eV 的势垒;2) Se 空位(VSe)的存在则会促进δ-InSe 的氧化反应,被氧化的过程仅需克服0.044 eV 的势垒,说明VSe 的存在使δ-InSe 在O2 环境下的稳定性显著下降,此外被O2 分子氧化的δ-InSe 单层有利于H2O 分子的解离吸附;3)含有In 空位(VIn)的δ-InSe被氧化的速率较慢,O2 分子在VIn 表面的物理吸附的吸附能和电荷转移与完美表面基本一致,被氧化的过程需克服1.234 eV 的势垒.这一研究结果将为更好地理解单空位缺陷对δ-InSe 单层的氧化行为提供理论指导,同时为高可靠二维δ-InSe 器件的实验制备提供参考.
1 引言
作为一种新型蜂窝状结构层状材料,InSe 因其优异的光电性质,在电子和光电子器件领域具有极为广泛的应用前景[1,2].目前实验中常见的InSe 主要有3 种已知相,分别为β(D3h),γ(D3h)和ε(D3h)[3–5],均由同种单层结构(D3h)按照不同的层间堆积方式构成.2020年,Zhang 等[6]结合人工智能粒子群优化算法与第一性原理计算,从理论上设计出具有中心反演对称性的D3d单层结构构成的二维InSe半导体,分别是δ(D3d),ω(D3d)和ϕ(D3d),空间群分别为,P63mc,,并且均具有良好的动力学和热力学稳定性.与D3h所构成的3 种已知相相比,δ(D3d)-InSe 表现出更宽的带隙变化范围和更高的电子迁移率[6].
二维半导体材料的环境稳定性是其在器件应用中必须考虑的关键问题[7–11].例如,Gao 等[12]发现MoS2晶体管的性能易受空气中活性物质的影响,当MoS2晶体管暴露在空气中一个月后,在对其栅极和漏极施加电压时,漏极电流降低了2 个数量级.部分二维材料中的空位缺陷是导致其在空气环境中不稳定的主要因素.例如,Kc 等[13]发现完美MoS2单层在空气中是惰性的,由于S 空位缺陷的产生,导致其单层易被氧化;Guo 等[14]发现完美GaSe 在O2环境中具有较高的抗氧化性,而含有Se 空位缺陷的GaSe 易受O2分子影响.有报道显示,少层InSe 在空气中可以短时间内保持环境稳定,但暴露在空气中一个月后,光致发光(photoluminescence,PL)光谱出现明显信号减弱或消失[15].关于δ-InSe 的环境稳定性研究目前未见报道.因此,研究δ-InSe 在O2环境下的稳定性对于研究和发展InSe 半导体材料器件具有极其重要的意义.
本文将采用基于密度泛函理论的第一性原理方法,研究二维δ-InSe 材料在O2环境下的稳定性.研究结果将为深入了解δ-InSe 的氧化行为提供有价值的参考,同时,为进一步推进二维δ-InSe半导体材料在电子、光电子等领域的应用进程提供研究思路.
2 计算方法
本文第一性原理计算基于密度泛函理论(density functional theory,DFT)[16,17]的VASP(Viennaab initioSimulation Package)[18]软件包.采用含有广义梯度近似(generalized gradient approximation,GGA)下的Perdew-Burke-Ernzerhof(PBE)泛函描述电子交换关联泛函[19–21].采用投影缀加平面波(projector-augmented wave,PAW)方法描述离子实和价电子的相互作用[22].采用DFT-D3 方法描述O2分子吸附在InSe 表面的长程范德瓦耳斯相互作用[23].平面基组的截断能设置为450 eV,能量收敛判据为10–5eV/Å (1 Å=10–10m),力收敛判据为0.02 eV/Å.真空层厚度设置为20 Å,避免周期性重复导致的相互作用.布里渊区积分采用Monkhorst-Pack[24]型网格,k点取值2×2×1.使用CI-NEB (climbing-imageNudged elastic band)[25]方法寻找不同状态之间转换的最小能量路径以及势垒.
为研究O2分子在δ-InSe 表面最稳定的吸附构型,本文所用到的吸附能计算表达式为[26]
其中,EInSe+O2表示完美或含有空位缺陷的δ-InSe单层的能量;EInSe表示O2分子物理吸附在δ-InSe单层的能量;EO2为O2分子的能量.根据定义,Ead负(正)值表示吸附过程为放(吸)热.
为更好地理解O2分子和δ-InSe 单层之间的相互作用和相应的电子转移,引入了差分电荷密度的计算[27]:
其中,ρAB为O2分子吸附在δ-InSe 单层的电荷密度;ρA和ρB分别为O2分子以及δ-InSe 的电荷密度.
3 结果与讨论
3.1 完美δ-InSe 单层的稳定性
本文构建的δ-InSe 是单层4×4×1 超胞结构.如图1(a)所示,该构型在层间方向上具有Se-In-In-Se 的原子层顺序,其空间群为,晶格常数为a=b=4.08 Å,Se—In 键长为2.68 Å,In—In键长为2.78 Å,与文献[6]中报道的结果一致.完美δ-InSe 单层的能带结构如图1(b)所示,其带隙大小为1.45 eV,高对称点路径为Γ—M—K—Γ,导带底(CBM)位于Γ点,价带顶(VBM)位于Γ 点和M点之间.图1(c),(d)为完美δ-InSe 单层的总态密度(TDOS)和分波态密度(PDOS),可以看出VBM 主要由Se 4p 和In 5p 态组成,CBM 主要由Se 4p 和In 5s 态组成.
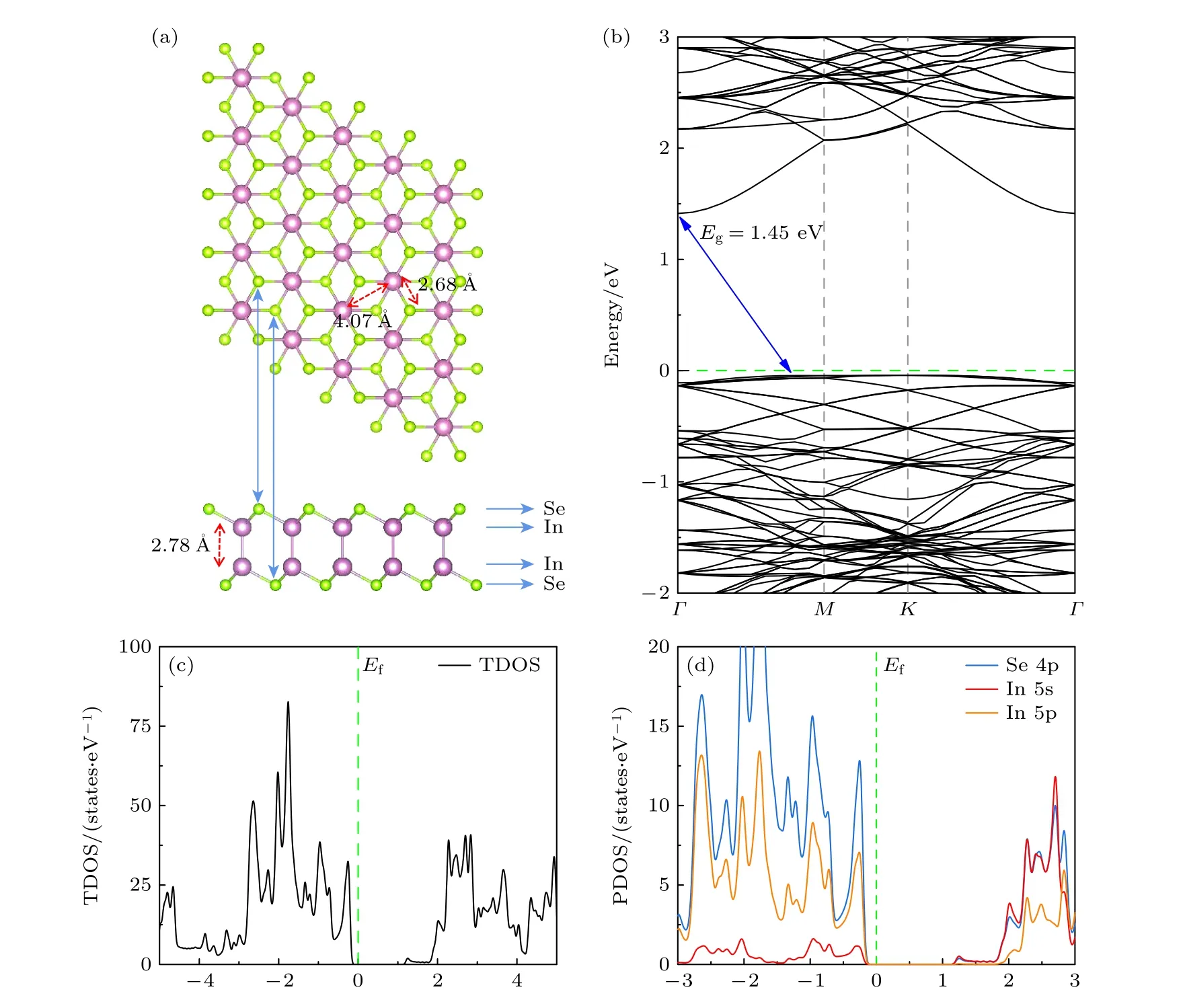
图1 完美δ-InSe 单层的晶体结构、能带结构和态密度图(a) 完美δ-InSe 单层超胞结构(俯视图和侧视图),绿色代表Se 原子,紫色代表In 原子;(b) 完美δ-InSe 单层的能带结构,蓝色实线箭头代表带隙(Eg),绿色虚线代表费米能级(Ef);(c),(d) 完美δ-InSe单层的TDOS 和PDOSFig.1.Crystal structure,band structure,and density of states diagram of perfect δ-InSe monolayer: (a) Supercell structure of perfect δ-InSe monolayer (top view and side view),where green represents Se atoms and purple represents In atoms;(b) band structure of perfect δ-InSe monolaye,where the blue solid arrow represents the band gap (Eg) and the green dashed line represents the Fermi level (Ef);(c),(d) TDOS and PDOS of perfect δ-InSe monolayer.
图2 考虑了O2分子物理吸附在δ-InSe 单层表面不同的吸附位点[14],其中包括In 原子顶部(TIn),上层Se 原子顶部(TSe1),下层Se 原子顶部(TSe2)和Se—Se 桥位顶部(TB).同时,对于每个吸附位点构建了两种分子取向,即O2分子平行(O2//δ-InSe)或垂直(O2⊥δ-InSe)于δ-InSe 单层表面.表1 列出了每一种构型的吸附能(Ead)和吸附距离(have),have为两个O 原子与δ-InSe 表面最短距离的平均值.根据所有构型的吸附能,发现O2分子最稳定的吸附构型为图2(a),其吸附能为–0.078 eV,吸附距离为3.19 Å,O—O 键键长保持不变(1.23 Å).因此,可以说明O2分子物理吸附在完美δ-InSe 表面,并且完美δ-InSe 表面具有良好的惰性.
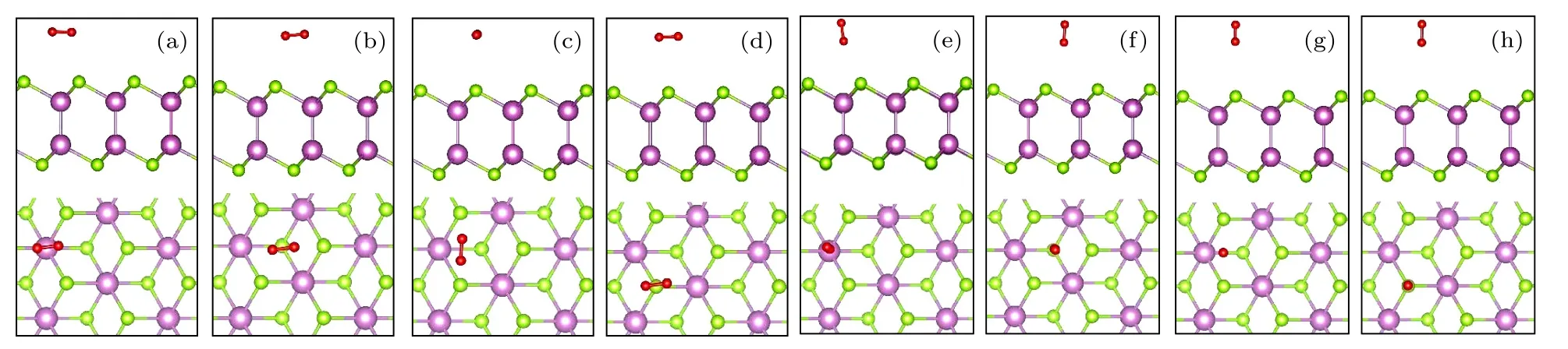
图2 O2 分子在完美δ-InSe 单层表面的不同吸附位点(侧视图和俯视图),红色代表O 原子(a),(e) TIn;(b),(f) TSe2;(c),(g) TB;(d),(h) TSe1Fig.2.Different adsorption sites of O2 molecules on the perfect δ-InSe monolayer surface (side view and top view),with red representing O atoms: (a),(e) TIn;(b),(f) TSe2;(c),(g) TB;(d),(h) TSe1.

表1 O2 分子在完美δ-InSe 单层表面吸附的吸附能(Ead)和吸附距离(have)Table 1.Adsorption energy (Ead) and adsorption distance (have) of O2 molecules on perfect δ-InSe monolayer surface.
为进一步研究完美δ-InSe 单层的稳定性,分析了O2分子在完美δ-InSe 单层表面的物理吸附到解离吸附的反应途径,如图3 所示.O2分子物理吸附在δ-InSe 单层表面(初态(IS)),吸附距离为3.30 Å,O—O 键键长为1.23 Å.随着O2分子不断接近表面,过渡态(TS)的O—O 键断裂,其中一个O 原子吸附在Se 原子上,另一个则吸附在In 原子上.末态(FS)的O 原子分别插入到两侧的In—Se键中形成In—O—Se键,所形成的Se—O 键和In—O 键长分别为1.77 Å和2.12 Å.O2分子从物理吸附到解离吸附的过程需要克服1.827 eV 的势垒,较高的势垒表明其在O2环境中被氧化的速率非常慢.
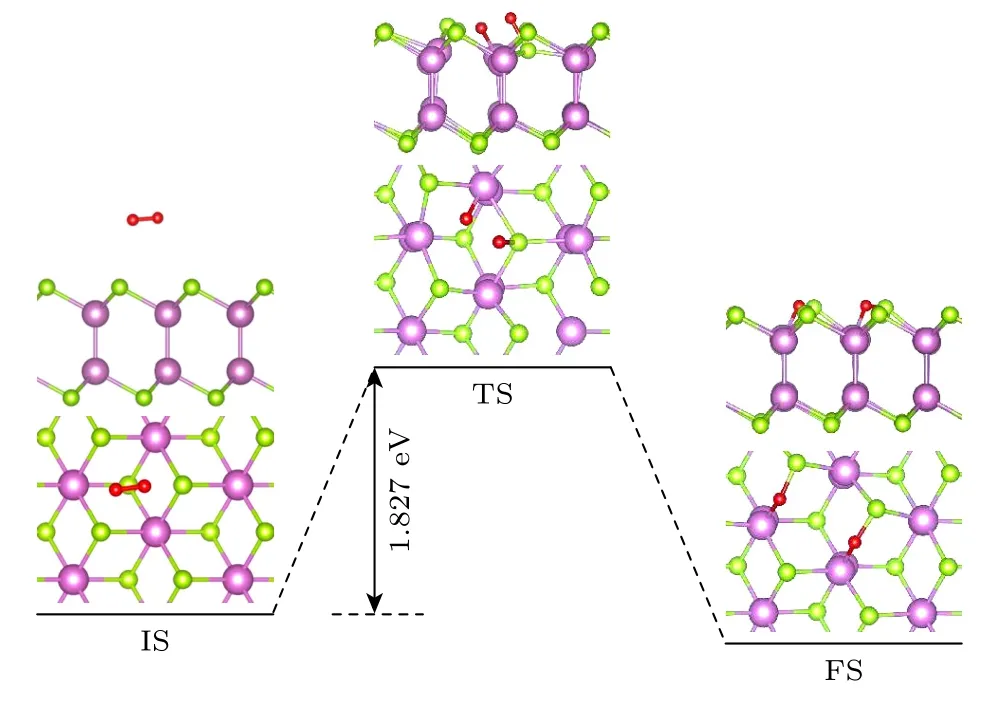
图3 O2 分子在完美δ-InSe 单层上解离成两个O 原子的反应途径Fig.3.Reaction pathway for an O2 molecule to dissociate into two O atom on perfect δ-InSe monolayer.
3.2 含有单空位缺陷δ-InSe 单层的稳定性
图4(a)为含有VSe缺陷的δ-InSe 单层(δ-InSe-VSe)超胞结构,围绕VSe的3 个上层In 原子向VSe中心发生偏移,导致周围的Se—In 键和In—In键分别拉伸至2.72 Å和2.83 Å.图4(b)为δ-InSe-VSe的能带结构,其带隙大小为1.02 eV,与完美δ-InSe 单层带 隙相比 减小了0.43 eV.CBM位于M点,VBM 位于K点.图4(c),(d)为δ-InSe-VSe的TDOS 和PDOS,与完美δ-InSe 的态密度(见图1(c))相比,可以看出在VBM 和CBM 附近产生了缺陷峰.该缺陷峰 在VBM 附近(–0.3 到–0.1 eV),峰宽为0.3 eV,主要由Se 4p 和In 5p 态组成;在CBM 附近(0.8—1.2 eV),峰宽为0.6 eV,主要由Se 4p,In 5s 和In 5p 态组成.
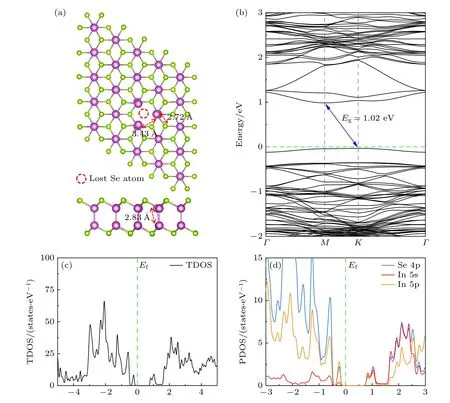
图4 δ-InSe-VSe 的晶体结构、能带结构和态密度图(a) δ-InSe-VSe 晶体结构(俯视图和侧视图);(b) δ-InSe-VSe 的能带结构,蓝色实线箭头代表带隙(Eg),绿色虚线代表费米能级(Ef);(c),(d) δ-InSe-VSe 单层的TDOS 和PDOSFig.4.Crystal structure,band structure,and density of states diagram of δ-InSe-VSe: (a) Crystal structure diagrams of δ-InSe-VSe(top view and side view);(b) band structure of δ-InSe-VSe,where the blue solid arrow represents the band gap (Eg) and the green dashed line represents the Fermi level (Ef);(c),(d) TDOS and PDOS of δ-InSe-VSe.
含有VIn缺陷的δ-InSe 单层(δ-InSe-VIn)超胞结构如图5(a)所示,VIn底部的In 原子向上偏移,而VIn顶部的3 个未配位的Se 原子向下偏移,且与顶部的In 原子形成Se—In 键(键长为2.87 Å).根据δ-InSe-VIn的能带结构和态密度(图5(b)—(d)),可以看出在费米能级(Ef)附近出现了明显的缺陷峰,自旋向上和自旋向下的自旋态劈裂.
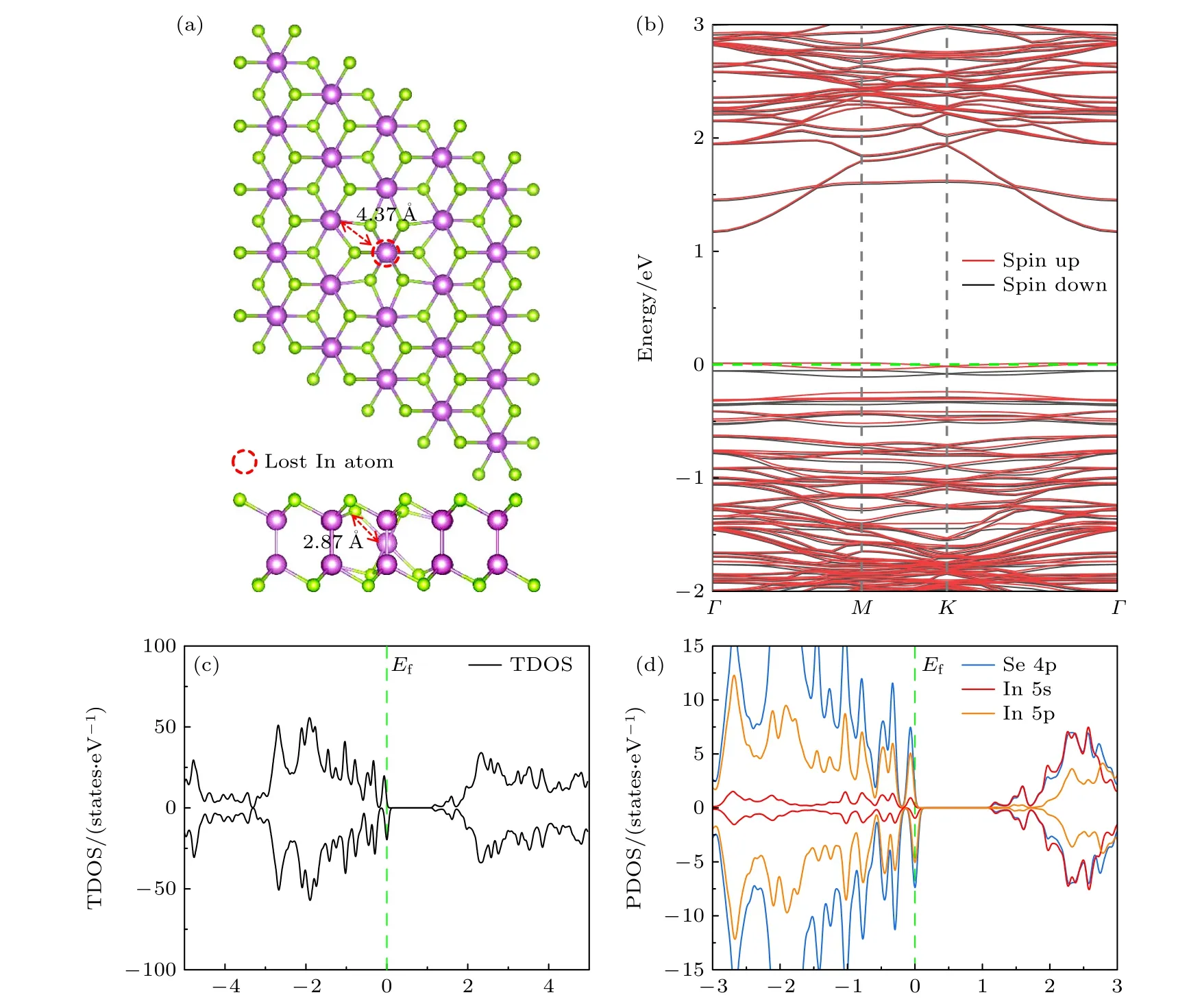
图5 δ-InSe-VIn 的晶体结构、能带结构和态密度图(a) δ-InSe-VIn 晶体结构(俯视图和侧视图);(b) δ-InSe-VIn 的能带结构;(c),(d) δ-InSe-VIn 单层的TDOS 和PDOSFig.5.Crystal structure,band structure,and density of states diagram of δ-InSe-VIn: (a) Crystal structure diagrams of δ-InSe-VIn(top view and side view);(b) band structure of δ-InSe-VIn;(c),(d) TDOS and PDOS of δ-InSe-VIn.
图6 为O2分子在含有单空位缺陷的δ-InSe单层表面上的物理吸附构型.图6(a),(b)为O2分子在δ-InSe-VSe表面的两种吸附构型,图6(c),(d)为O2分子在δ-InSe-VIn表面的两种吸附构型.根据表2 可知,O2分子吸附在δ-InSe-VSe和δ-InSe-VIn最稳定的构型分别为图6(a),(c),其吸附能分别为–0.152 eV 和–0.093 eV,与完美δ-InSe 单层相比(图2(a)),吸附能分别减小了0.074 eV 和0.015 eV.O2分子吸附在δ-InSe-VSe表面的吸附能的变化更加显著,吸附在δ-InSe-VIn表面的吸附能的变化较小,这表明在O2环境下,VSe易受O2分子影响.
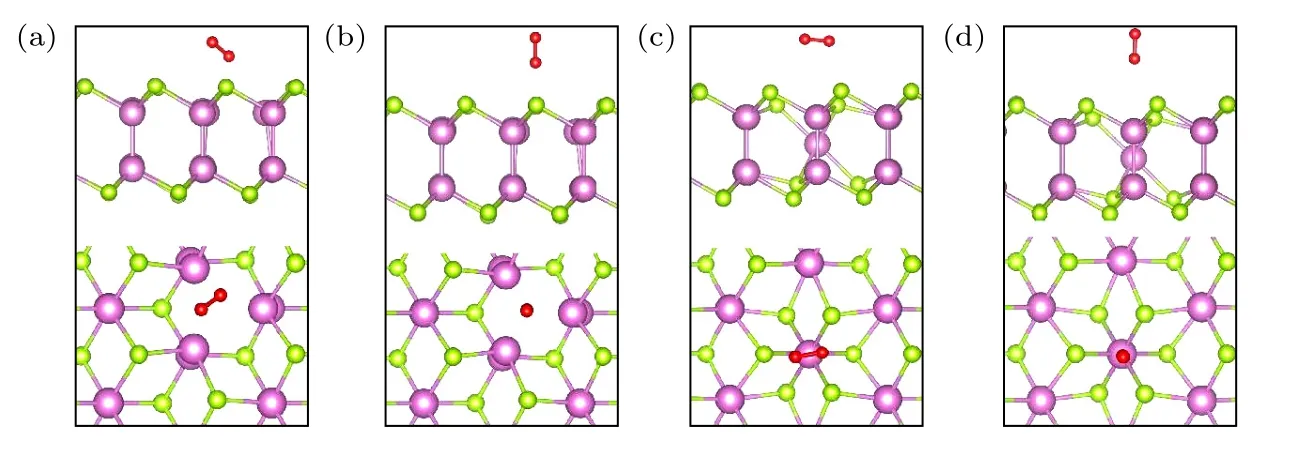
图6 O2 分子在δ-InSe-VSe 和δ-InSe-VIn 的吸附位点(侧视图和俯视图)(a) TVSe-1;(b) TVSe-2;(c) TVIn-1;(d) TVIn-2Fig.6.Adsorption sites of O2 molecules on the δ-InSe-VSe and δ-InSe-VIn (top view and side view): (a) TVSe-1;(b) TVSe-2;(c) TVIn-1;(d) TVIn-2.

表2 O2 分子在δ-InSe-VSe 和δ-InSe-VIn 表面的吸附能(Ead)和吸附距离(have)Table 2.Adsorption energy (Ead) and adsorption distance (have) of O2 molecules on δ-InSe-VSe and δ-InSe-VIn surfaces,respectively.
为研究O2分子与单层表面相互作用的过程中电子的移动与再分布[28],分析了其差分电荷密度.图7(a)为O2分子吸附在完美δ-InSe 单层表面(吸附位点为TIn)的差分电荷密度图,O2分子的吸附引起了微弱的电荷转移,电荷聚集区域主要集中在O2分子附近.Bader 电荷结果表明,O2分子获得了0.019e的电荷转移量.图7(b)为O2分子在δ-InSe-VSe表面(吸附位点为TVSe-1)的差分电荷密度图,可以看出O2分子的吸附引起的电荷转移比完美表面更为明显,电荷聚集区域主要集中在O2分子周围,电荷损耗区域主要集中在δ-InSe单层表面,O2分子获得了0.136e的电荷转移量.O2分子在δ-InSe-VIn表面(吸附位点为TVIn-1)的差分电荷密度图,如图7(c)所示,O2分子获得了0.023e的电荷转移量,与完美表面相比仅相差0.004e.结果表明,O2分子与δ-InSe-VSe表面的相互作用更为显著,而与δ-InSe-VIn表面的相互作用与完美表面基本一致.

图7 O2 分子吸附在δ-InSe 单层的差分电荷密度,黄色部分表示电荷积累区域,蓝色部分表示电荷损耗区域(等值面设为1.5×10–4 e/Bohr3)(a) O2 分子在完美δ-InSe 的差分电荷密度;(b) O2 分子在δ-InSe-VSe 的差分电荷密度;(c) O2 分子在δ-InSe-VIn 的差分电荷密度Fig.7.Differential charge density of O2 adsorbed on δ-InSe monolayer,where yellow regions indicate charge accumulation and blue regions indicate charge depletion (the equivalent surface is set to 1.5×10–4 e/Bohr3) : (a) Differential charge density of O2 adsorbed on perfect δ-InSe;(b) differential charge density of O2 adsorbed on δ-InSe-VSe;(c) differential charge density of O2 adsorbed on δ-InSe-VIn.
图8 为O2分子在δ-InSe-VSe表面从物理吸附到解离吸附的反应途径.当δ-InSe 单层表面存在VSe时,O2分子首先物理吸附在VSe上方1.83 Å的位置,其O—O 键键长为1.23 Å.当O2分子到达单层表面上方1.08 Å的高度(TS1),O—O 键键长被拉伸至1.27 Å.随后,O2分子化学吸附到VSe处(MS),与VSe周围3 个未配位的In 原子成键.在接下来的反应步骤中(TS2),O 原子发生偏移,O—O 键长被拉伸至1.51 Å.末态(FS) O—O 键断裂,其中一个O 原子化学吸附在VSe处,所形成的In—O 键键长为2.20 Å,另一个O 原子进入到周围In—In 键中,形成In—O—In 键.
研究结果表明,当δ-InSe 单层表面含有VSe时,O2分子从物理吸附到化学吸附的势垒仅为0.044 eV,与完美δ-InSe 单层相比,势垒降低了1.783 eV.此外,O2分子从化学吸附到解离吸附的势垒仅为0.001 eV,表明VSe在O2环境中则会促进δ-InSe 的氧化反应.
图9 为O2分子在δ-InSe-VIn表面从物理吸附到解离吸附的反应途径.当δ-InSe 单层表面存在VIn时,O2分子首先物理吸附在VIn上方2.63 Å的位置,其O—O 键键长1.23 Å.随后,O 原子与VIn周围的两个Se 原子形成Se—O 键(TS),O—O 键被拉伸至1.64 Å.在末态(FS)下,O—O 键断裂.对于含有VIn的δ-InSe 单层,O2分子从物理吸附到解离吸附需要克服1.234 eV 的势垒,与完美δ-InSe单层相比,势垒仅降低了0.593 eV.较高的势垒表明,在O2环境下δ-InSe-VIn被氧化速率较慢.
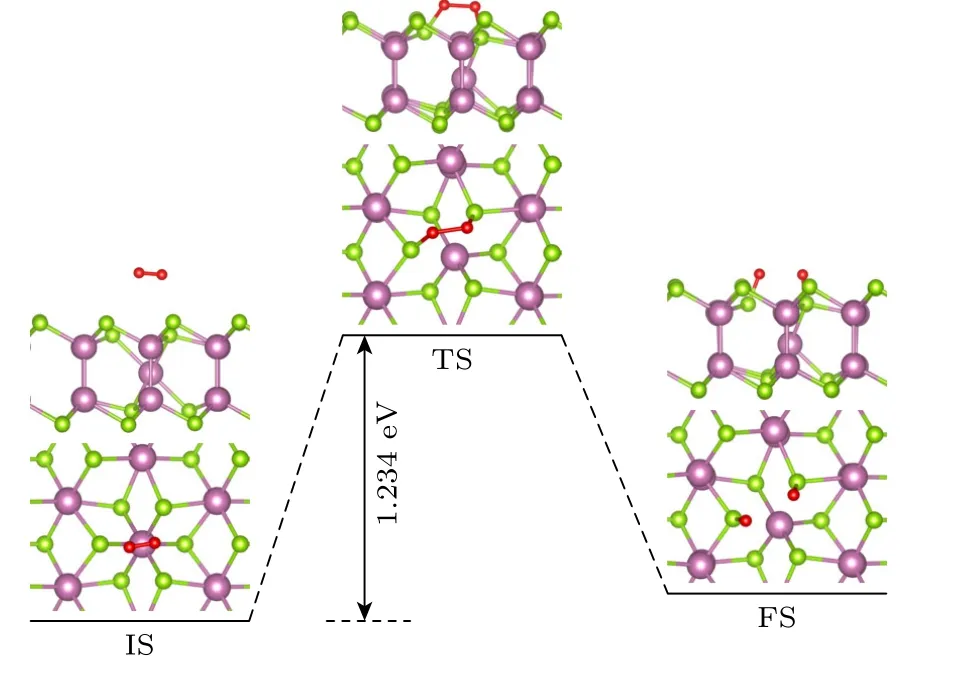
图9 O2 分子在δ-InSe-VIn 解离成两个O 原子的反应途径Fig.9.Reaction pathway for an O2 molecule to dissociate into two O atom on δ-InSe-VIn.
由于Se 空位的存在,δ-InSe 单层易于被O2分子氧化,因此,在此基础上继续考虑H2O 分子在被O2氧化的δ-InSe 单层上发生解离的反应途径.从图10 可以看出,H2O 分子首先物理吸附在被氧化的δ-InSe 单层上方1.54 Å的位置.随后,H2O 分子化学吸附在被氧化的δ-InSe 单层上.在末态(FS)下,H2O 分子的一个O—H 键断裂,一个羟基与In 原子成键,另一个H 原子与表面的O 原子成键.从物理吸附到解离吸附的过程中,H2O 分子在该表面所需要克服的势垒仅为0.136 eV.因此,表明被O2分子氧化的δ-InSe 单层有利于H2O 分子的解离吸附.
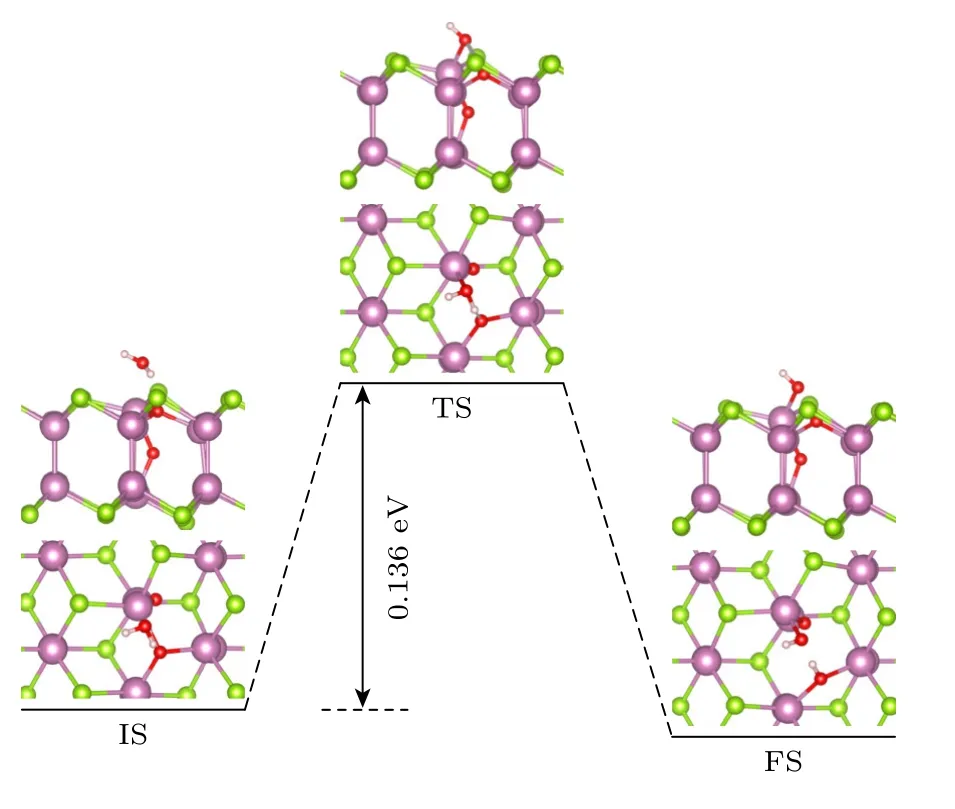
图10 H2O 分子 在被O2 氧化的δ-InSe 单层上发生 解离的反应途径Fig.10.Dissociation pathway of H2O molecules on the δ-InSe monolayer oxidized by oxygen.
4 结论
本文采用基于密度泛函理论的第一性原理方法,系统地研究了完美和含有单空位缺陷δ-InSe在O2环境下的稳定性问题.结果表明,O2分子在完美δ-InSe 表面的吸附能仅有–0.078 eV,表现为物理吸附.O2分子在其表面从物理吸附到解离吸附需要克服1.827 eV 的势垒,说明完美δ-InSe 表现出较高的抗氧化能力.然而,VSe的存在则会促进δ-InSe 的氧化反应,被氧化的过程仅需克服0.044 eV 的势垒,此外,H2O 分子在被O2分子氧化的δ-InSe 单层表面上,从物理吸附到解离吸附需要克服0.136 eV 的势垒.相比之下,含有VIn的δ-InSe 单层被氧化的速率较慢,O2分子在VIn表面的物理吸附的吸附能和电荷转移与完美表面相似,被氧化的过程需克服1.234 eV 的势垒.
综上所述,在O2环境下,Se 空位是导致二维δ-InSe 材料不稳定的主要因素,并且被O2分子氧化的δ-InSe 单层有利于H2O 分子的解离吸附.相比于D3h单层结构所构成的相,含有Se 空位的δ-InSe 的稳定性稍差[9,11,29].因此,在未来二维δ-InSe 的材料的制备和使用过程中,保护措施尤为重要.基于上述研究结果,对于提高二维δ-InSe 在实验中的稳定性有以下三点建议: 首先,在材料制备过程中,可以通过富硒的生长环境来减少硒空缺的形成;其次,在材料使用过程中,可以采用化学手段钝化或填补硒空缺,进一步提升二维δ-InSe的环境稳定性;最后,在材料的使用过程中,尽量避免潮湿的环境.以上研究结果为理解单空位缺陷对δ-InSe 单层的氧化行为提供理论指导,同时为高可靠二维δ-InSe 器件的实验制备提供参考.
猜你喜欢
分子催化(2022年1期)2022-11-02
电子制作(2019年15期)2019-08-27
陶瓷学报(2019年5期)2019-01-12
测控技术(2018年9期)2018-11-25
上海大中型电机(2017年4期)2017-02-06
电子制作(2017年19期)2017-02-02
晋中学院学报(2015年3期)2015-04-01
读者欣赏(2014年6期)2014-07-03
郑州大学学报(理学版)(2014年4期)2014-03-01
语文知识(2014年2期)2014-02-28
