
阿普斯特合成路线图解
2024-03-14 09:06叶琼仙罗统有邓吉聪谭志聪
江西化工 2024年1期
叶琼仙,罗统有,邓吉聪,谭志聪
(1.广东华南药业集团有限公司,广东东莞,523320;2.广东先强药业有限公司,广东广州,510900)
0 引言
阿普斯特(Apremilast,1),化学名为(S)-2-[1-(3-乙氧基-4-甲氧基苯基)-2-甲磺酰基乙基]-4-乙酰基氨基异吲哚啉-1,3-二酮,是美国Celgene 公司研发的一种口服类选择性磷酸二酯酶4 抑制剂,分别获得FDA、EMA 监管机构批准用于活动性银屑病关节炎(PsA)和中度至重度斑块型银屑病的治疗。
2014 年,该药首先在美国获准上市,商品名为Otezla,是FDA 首个批准用于斑块型银屑病(牛皮癣)及银屑病关节炎治疗的口服制剂。阿普斯特的对环单磷酸腺苷(cAMP)特异性,可使细胞内cAMP 水平上升,调节胞内促炎与抗炎因子作用,然后与抗炎因子作用,抑制参与银屑病发病机制中的多个炎症的活性,减轻关节肿胀并改善关节部位的生理机能,可有效治疗银屑病[1]。
1 阿普司特药理作用
阿普司特是炎症疾病的潜在口服治疗药物,能够对TNF-α,IL-2 及其他多种促炎性介质(PDE4,IL-2、干扰素γ、白三烯、NO 合成酶)生成产生抑制性,其药理作用是利用抑制PDE4 酶增强细胞内环单磷酸腺苷水平特性,增强IL-10 等抗炎细胞因子,从而对多条炎症通路产生影响。患者在使用阿普司特药物后,能够快速抑制肿瘤坏死因子(TNF-α)及其他炎症细胞因子的表达,达到较好的抗炎目的。
在一系列的临床研究中,该药物表现出良好的治疗效果,同时具备较高的安全性和耐受性。Ⅱ期临床试验研究表明,患者在服用阿普司特药物后,银屑病损伤面可减少75%以上,病情严重程度明显降低,且仅存轻微不良反应;Ⅲ期临床试验研究表明,该药物总体耐受性良好,安全性较强。
2014 年3 月21 日,该药物率先获得美国食品药品监督管理局(FDA)批准上市,现已成为治疗成人活跃型银屑病性关节炎的关键药物。
2 合成路线分析
阿普斯特(1)经逆合成分析,可通过S-1-(3-乙氧基-4-甲氧基苯基)-2-甲磺酰基乙胺(2)和3-乙酰氨基邻苯二甲酸酐(3)胺化而得(图1)。本文对阿普斯特(1)的合成路线进行了综述(图2)。

图1 阿普斯特逆合成分析
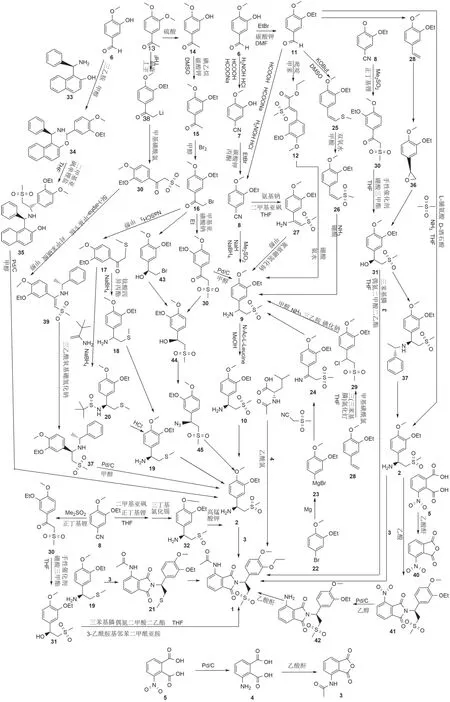
图2 阿普斯特合成路线图解
2.1 拆分法
A 法:以3-乙氧基-4-甲氧基苯腈(6)为原料,经盐酸羟胺缩合得到(7),溴乙烷取代合成(8),加成还原得(9),经N-乙酰基L-亮氨酸手性拆分得到(2),和(3)胺化得到(1)[2]。
B 法:以3-乙氧基-4-甲氧基苯甲醛(11)为原料,经加成成烯得(12),加成得(9),经拆分得到(2),和(3)胺化得到(1)[3]。
C 法:以3-乙氧基-4-甲氧基苯甲醛(11)为原料,经加成成腈(8),加成还原得(9),经拆分得到(2),和(3)胺化得到(1)[4]。
D 法:以3-甲氧基-4-甲氧基苯乙酮(13)为原料,经酸解得(14),醚化成(15),取代得(16),硫醚化得(17),还原胺化得(18),经化学拆分得(19),再氧化成(2),和(3)胺化得到(1)[5]。
E 法:以3-甲氧基-4-甲氧基苯乙酮(13)为原料,经酸解得(14),醚化成(15),取代得(16),氧化得(30),还原胺化得(9),经拆分得到(2),和(3)胺化得到(1)[6]。
F 法:1-(3-乙氧基-4-甲氧基苯基)-2-(甲基磺酰基)乙胺(9)经拆分剂成盐得到(10),和(4)缩合,再乙酰化得到(1),和(3)胺化得到(1)[7]。
G 法:以4-溴-2-乙氧基-1-甲氧基苯(22)为原料,先制备格式试剂得(23),再格氏反应得(24),还原得(9),经拆分得到(2),和(3)胺化得到(1)[8]。
H 法:以3-乙氧基-4-甲氧基苯甲醛(11)为原料,经加成成(25),氧化得(26),加成得(9),经拆分得到(2),和(3)胺化得到(1)[9]。
I 法:以3-甲氧基-4-甲氧基苯腈(8)为原料,成烯胺得(27),再还原成(9),经拆分得到(2),和(3)胺化得到(1)[10]。
1.8 统计学处理 采用SPSS 19.0统计软件进行资料处理。正态分布的资料采用算术均数和标准差描述其分布特征,组间比较采用独立样本t检验;非正态分布的资料采用中位数和四分位间距进行描述,组间比较采用非参数检验(Mann-Whitney U检验)。计数资料采用百分数表示,组间比较采用常规或校正卡方检验。此外采用logistics回归分析患者术后1年生存的影响因素。检验水准a=0.05。
J 法:以3-甲氧基-4-甲氧基苯乙烯(28)为原料,加成得(29),取代得(9),经拆分得到(2),和(3)胺化得到(1)[11]。
该合成路线所用原料便宜易得,中间体分离简便,反应操作安全,三废处理容易,且反应过程中所用设备简单,无需采用特殊设备及器材,更不用高压、高真空。大量应用实践表明,该合成路线总收率较高,生产成本低,满足工业化生产要求。
2.2 不对称合成法
K 法:以3-甲氧基-4-甲氧基苯乙酮(13)为原料,经酸解得(14),醚化成(15),取代得(16),硫醚化得(17),还原胺化得(18),不对称合成得(20),酸解得(19),再氧化成(2),和(3)胺化得到(1)[5]。
L 法:以3-乙氧基-4-甲氧基苯腈(6)为原料,经盐酸羟胺缩合得到(7),溴乙烷取代合成(8),加成得(30),经不对称还原得(31),和(3)胺化得到(1)[12]。
M 法:以3-甲氧基-4-甲氧基苯腈(8)为原料,加成得(32),再经氧化得(2),和(3)胺化得到(1)[13]。
N 法:以3-乙氧基-4-甲氧基苯腈(6)为原料,加成环合得(34),加成得(35),氢解得(2),和(3)胺化得到(1)[14]。
O 法:以3-乙氧基-4-甲氧基苯甲醛(11)为原料,成烯得(28),接着不对称加成得(36),开环加成得(31),上苄胺得(37),氢解得(2),和(3)胺化得到(1)[15]。
P 法:以3-甲氧基-4-甲氧基苯乙酮(13)为原料,正丁基锂作用下成锂化合物(38),格氏反应后得(30),经不对称还原胺化得(39),还原得(37),氢解得(2),和(3)胺化得到(1)[16]。
Q 法:以3-乙氧基-4-甲氧基苯甲醛(11)为原料,经不对称加成得(2),和(3)胺化得到(1)[17]。
R 法:以3-硝基-邻苯二甲酸(5)为原料,经乙酸酐环合得(40),和(2)胺化得(41),氢化还原得(42),再乙酰化得(1)[18]。
S 法:以(15)为起始物料,取代得(16),经不对称还原得(43),取代得(44),重氮化得(45),再还原得(2),和(3)胺化得到(1)[19]。
T 法:以(15)为起始物料,取代得(16),氧化得(30),经不对称还原得(44),重氮化得(45),再还原得(2),和(3)胺化得到(1)[19]。
该合成路线步骤较少,且在胺化处理时会用到四乙氧基钛,其遇水或水蒸气会发生反应而放出有毒或易燃气体,为后续处理带来较大难度,同时,所得到手性中间体ee 值仅为 80%左右,仍需进一步拆分。
3-乙氧-4-甲氧基苯腈酯(1)与二甲基磺胺(2)进行加成制备希夫碱(2),通过酸解制得产物(3),在不对称催化下进行加氢还原制得R 构型中间体(4),与4-硝基苯二甲酰亚胺酯(4)发生 Mitsunobu 反应,生成化合物(5), 再经钯炭氢化还原、乙酰化,最终实现阿普司特的合成[20-22]。
这条合成路线虽然只需6 步就可直接合成阿普司特,但需用到昂贵的手性催化剂,且 Mitsunobu 反应为无水无氧反应,反应制备操作要求严格,对生产设备要求较高,因而很难实现规模化生产。
3 结语
本研究基于阿普司特药理作用,对该药物合成的拆分法、不对称合成法两种路线进行详细论述,但仍有部分研究未能完成,仍需进一步深入研究。从理论上讲,对直接合成的手性化合物进行化学拆分,可以获得相当于手性化合物一半数量级的拆分产物,但在拆分环节,尽管通过优化拆分工艺,拆分收率有所提升,但因两个异构体在拆分系统中均存在不同程度的溶解性,导致拆分体系的母液中仍存在部分S-手性中间体I 和所有R构型中间产物 I,因此,需通过对R 构型混合产物进行消旋化处理,从而实现对该手性混合产物的有效拆分。现有的合成路线均采用正丁基锂作为原料,存在价格昂贵、安全隐患大、对工业设备要求高的问题,希望通过不断研究,能开发出药物合成的新思路和方法,为实现工业化生产提供便利。
猜你喜欢
青年文摘(彩版)(2022年4期)2022-12-29
青春(2019年1期)2019-10-17
铜仁学院学报(2018年6期)2018-07-05
壹读(2018年2期)2018-03-09
小星星·作文100分(2018年1期)2018-01-29
中成药(2017年4期)2017-05-17
中国洗涤用品工业(2016年2期)2016-02-28
中国塑料(2015年2期)2015-10-14
云南中医学院学报(2015年2期)2015-07-31
华东理工大学学报(自然科学版)(2014年1期)2014-02-27
