高效液相色谱法测定新型促性腺激素释放激素拮抗剂LPM7100328原料药中的有关物质
2024-03-14 03:48李丽霞王旭东马树芝闫娜娜周凤梅车鑫烟台大学药学院山东烟台264005烟台大学分子药理和药物评价教育部重点实验室山东烟台264005山东绿叶制药有限公司长效和靶向制剂国家重点实验室山东烟台264670
中南药学 2024年2期
李丽霞,王旭东,马树芝,闫娜娜,,周凤梅,车鑫,*(1.烟台大学药学院,山东 烟台 264005;2.烟台大学分子药理和药物评价教育部重点实验室,山东 烟台 264005;.山东绿叶制药有限公司长效和靶向制剂国家重点实验室,山东 烟台 264670)
促性腺激素释放激素(GnRH)是由下丘脑分泌的一种十肽,可通过反馈控制垂体分泌促卵泡素和促黄体素,特异性和选择性地作用于GnRH的拮抗剂或激动剂,可用于性激素依赖性失衡相关疾病的治疗,其用于前列腺癌的治疗一直备受关注[1]。前列腺癌是男性常见恶性肿瘤之一,其发病率和死亡率仅次于肺癌。雄激素剥夺治疗(ADT)是局部晚期、复发性和转移性前列腺癌标准治疗的关键组成部分,其中GnRH激动剂(如戈舍瑞林、亮丙瑞林等)的使用最为常见[2-4],但GnRH激动剂在结合受体初始会暂时性促进促性腺激素的分泌,进而存在短暂性病情恶化的可能性,必要时还需联用抗雄激素药物[5];与激动剂不同,GnRH拮抗剂立即快速可逆性地抑制促性腺激素的分泌[6],可避免“点火效应”的发生[7],降低心血管疾病的发生风险[8]。目前有两种GnRH拮抗剂适用于晚期前列腺癌的治疗,分别是地加瑞克(注射用多肽药物)[9]和瑞卢戈利(Relugolix)(口服活性非肽类小分子药物)[10-11]。在研的具有GnRH拮抗活性的化合物多为肽类化合物,鉴于肽类GnRH拮抗剂存在口服吸收性、稳定性差等相关问题,仍需要继续研究开发临床效果更佳的口服非肽类小分子GnRH拮抗剂。
LPM7100328是山东绿叶制药有限公司针对瑞卢戈利进行结构改造,设计合成的新化合物,是一种新型的口服小分子GnRH受体拮抗剂,可用于晚期前列腺癌的治疗[12]。其结构式见图1。LPM7100328于药物研发早期的体外及动物实验表明,其对人源GnRH受体有明显的结合能力和抑制作用,与Relugolix相比,具有更高的口服生物利用度以及更小的心脏毒性。作为正处于Ⅰ期临床研究的1类新药,LPM7100328具有良好的临床前景。

图1 LPM7100328化学结构式Fig 1 Chemical structure of LPM7100328
在LPM7100328合成过程中,参与反应的物料以及引入的副产物未能在精制过程中随母液去除而残留在原料药中,即杂质A、B、C。本研究首次建立了LPM7100328原料药中有关物质检查的HPLC分析方法,并针对杂质A、B、C进行了全面的方法学验证,现报道如下。
1 仪器与试药
Agilent1260系列液相色谱仪(配有四元泵、自动进样器、柱温箱、紫外检测器、二极管阵列检测器和ChemStation工作站,美国Agilent公司);XS105电子天平、S400-K pH计(梅特勒-托利多公司);SK7210HP超声波清洗器(上海科导超声仪器有限公司)。
LPM7100328原料药(批号:2209281、2209282、2209283)、LPM7100328对照品(批号:2203301,纯度:99.2%)、杂质A对照品(批号:2200517,纯度:95.2%)、杂质B对照品(批号:2200621,纯度:98.8%)、杂质C对照品(批号:2204301,纯度:98.9%)(山东绿叶制药有限公司);乙腈、甲醇均为色谱纯;水为纯化水;其他试剂均为分析纯。
2 方法与结果
2.1 色谱条件
色谱柱:ACE Excel Super C18(4.6 mm×150 mm,3 µm),杂质捕集小柱:Welch C18(4.6 mm×50 mm);流动相A为0.01 mol·L-1磷酸二氢钾水溶液(用磷酸调节pH值至2.5),流动相B为甲醇-乙腈(20∶80);流速:1.0 mL·min-1;柱温:40℃;检测波长:248 nm;进样量:5 µL;梯度洗脱,洗脱程序见表1。

表1 梯度洗脱程序Tab 1 Gradient elution program
2.2 溶液配制
2.2.1 溶剂 以0.1%磷酸水溶液-乙腈(1∶1)为溶剂,将溶剂作为空白对照溶液。
2.2.2 对照品溶液 取LPM7100328对照品约12.5 mg,精密称定,置50 mL量瓶中,加适量溶剂溶解并稀释至刻度,摇匀,得对照品储备液;精密量取对照品储备液0.5 mL置50 mL量瓶中,加溶剂稀释至刻度,摇匀,即得。
2.2.3 杂质对照品储备液 分别取杂质A、B、C对照品约10 mg、15 mg和25 mg,精密称定,置50 mL量瓶中,加适量溶剂溶解并稀释至刻度,摇匀,即得。
2.2.4 系统适用性溶液 取LPM7100328对照品约25 mg,精密称定,置50 mL量瓶中,加适量溶剂溶解,加入杂质对照品储备液0.5 mL,加溶剂稀释至刻度,摇匀,即得。
2.2.5 供试品溶液 取LPM7100328原料药约25 mg,精密称定,置50 mL量瓶中,加适量溶剂溶解并稀释至刻度,摇匀,即得。
2.3 系统适用性试验
取“2.2”项下空白对照溶液和系统适用性溶液,进样测定,记录色谱图,见图2。空白对照溶液无干扰,系统适用性溶液中各色谱峰间分离度均大于1.5,各杂质均能得到良好分离。
2.4 强制降解试验
称取LPM7100328原料药(批号:2209281)约20 mg于10 mL量瓶中,共8份,分别进行以下试验。
① 未破坏:加适量溶剂超声溶解并稀释至刻度,摇匀,即得;② 酸破坏:加4 mL溶剂超声溶解,加1 mol·L-1盐酸溶液1 mL,80℃烘箱中放置30 min,冷却,用1 mol·L-1氢氧化钠溶液中和,加溶剂稀释至刻度,摇匀,即得;③ 碱破坏:加4 mL溶剂超声溶解,加0.25 mol·L-1氢氧化钠溶液1 mL,室温放置10 min,用0.25 mol·L-1盐酸溶液中和,加溶剂稀释至刻度,摇匀,即得;④ 氧化破坏:加适量溶剂超声溶解,加3.0%的过氧化氢溶液1 mL,用溶剂稀释至刻度,摇匀,80℃烘箱中放置24 h,冷却,即得;⑤ 固体高温破坏:封口置于80℃烘箱中,10 d后取出,冷却,加适量溶剂超声溶解并稀释至刻度,摇匀,即得;⑥ 液体高温破坏:加适量溶剂超声溶解,80℃烘箱中放置24 h,冷却,加溶剂稀释至刻度,摇匀,即得;⑦ 高湿破坏:开口置于相对湿度为92.5%的干燥器内,10 d后取出,加适量溶剂超声溶解并稀释至刻度,摇匀,即得;⑧ 光照破坏:在(4500±500)lx照度下放置10 d后取出,加适量溶剂超声溶解并稀释至刻度,摇匀,即得。
通过考察LPM7100328的降解途径及杂质峰(尤其是降解杂质)与主峰的分离度,验证该方法的专属性[13]。取上述破坏溶液进样分析,记录色谱图,结果见图3。LPM7100328在酸、碱、氧化和液体高温破坏条件下不稳定,在其他条件下具有一定的稳定性。在各降解条件下杂质峰及主峰均能得到良好的分离,主峰纯度均大于0.990;物料守恒均在98%~101%,表明该方法具有良好的专属性。

图3 LPM7100328强制降解试验色谱图Fig 3 HPLC chromatograms of forced degradation tests of LPM7100328
2.5 检测限与定量限试验
取杂质A、B、C对照品各约2.5 mg,精密称定,置100 mL量瓶中,加适量溶剂溶解并稀释至刻度,摇匀,作为定量限杂质储备液,取定量限杂质储备液和“2.2.2”项下对照品溶液配制定量限溶液(相当于0.05%供试品溶液浓度),平行配制6份,依法进样,计算信噪比(S/N)及峰面积RSD值,以S/N≥10为定量限,结果LPM7100328及杂质A、B、C的定量限分别为0.2486、0.2380、0.2468、0.2471 µg·mL-1。
取定量限溶液3 mL置10 mL量瓶中,加溶剂稀释至刻度,依法进样分析,记录色谱图,以S/N≥3为检测限,结果LPM7100328及杂质A、B、C的检测限分别为0.0746、0.0714、0.0740、0.0741 µg·mL-1,表明本方法灵敏度良好。
2.6 线性关系考察
分别配制LPM7100328及各杂质限度浓度为50%、100%、150%、200%和定量限浓度的线性溶液,进样测定,记录色谱图。以LPM7100328及杂质A、B、C的质量浓度(X)为横坐标,其对应的峰面积(Y)为纵坐标,进行线性回归,并按F=k主/k杂计算杂质A、B、C的校正因子,结果如表2所示。

表2 线性试验结果Tab 2 Linearity test
2.7 准确度试验
取LPM7100328原料药(批号:2209281)25 mg,精密称定,置50 mL量瓶,加适量溶剂溶解后,分别加入定量限杂质储备液0.5 mL、杂质对照品储备液0.5、0.75 mL,制成LOQ(n=3)、100%(n=6)、150%(n=3)各杂质限度水平的加标溶液,进样测定,计算平均回收率。杂质A、B、C的平均回收率范围为97.0%~107.1%,RSD为2.2%~4.7%,表明该方法准确度良好。
2.8 精密度试验
2.8.1 重复性试验 取“2.7”项下限度浓度为100%的6份准确度溶液作为重复性溶液,进样测定,计算各杂质及总杂含量的RSD。结果显示,杂质A、B、C及最大未知单杂含量的RSD分别为2.5%、0.90%、1.1%、0%,总杂含量的RSD为0.60%,表明该方法重复性良好。
2.8.2 中间精密度试验 两人分别取同一批LPM7100328原料药样品,按“2.7”项下方法平行配制6份100%浓度的加标供试品溶液,于同一实验室不同日期,按“2.1”项下色谱条件在不同仪器上分别进样测定,并计算各杂质及总杂含量的RSD。结果杂质A、B、C及最大未知单杂含量的RSD分别为8.4%、0.90%、1.8%、0%,总杂含量的RSD为1.3%,表明本方法可满足精密度要求。
2.9 稳定性试验
取LPM7100328原料药(批号:2209281)约25 mg,精密称定,置50 mL量瓶中,加适量溶剂溶解,加入杂质对照品储备液0.5 mL,加溶剂稀释至刻度,摇匀,即得加标供试品溶液;将加标供试品溶液和“2.2.2”项下对照品溶液室温下放置72 h,分别于0、12、24、48、72 h进样测定,记录色谱图,并计算LPM7100328及各杂质与0 h相比的峰面积回收率。结果表明,72 h内LPM7100328回收率在99.6%~100.6%,杂质A、B、C和未知单杂的回收率在98.5%~112.3%,且无其他杂质产生。表明对照品溶液及加标供试品溶液室温放置72 h内稳定。
2.10 耐用性试验
按“2.2.5”项下方法配制供试品溶液,分别考察适当调节流动相pH(±0.2)、柱温(±2℃)、有机相组成比例(±5%)以及不同仪器对LPM7100328与杂质分离效果及样品含量测定的影响。结果表明,在不同色谱条件下,各色谱峰间分离度均大于1.5,各有关物质的检出情况基本无变化,表明该方法耐用性良好。
2.11 样品含量测定
取3批LPM7100328原料药,按“2.2.5”项下方法配制供试品溶液,进样测定,记录色谱图,按加校正因子的主成分自身对照法计算杂质A、B、C含量,未知杂质含量按不加校正因子的主成分自身对照法计算,结果如表3所示。杂质A、B、C,其他最大单杂及总杂检出量均符合限度标准(杂质A限度为0.20%,杂质B限度为0.30%,杂质C限度为0.50%,其他单杂限度为0.50%,总杂限度为1.5%)。
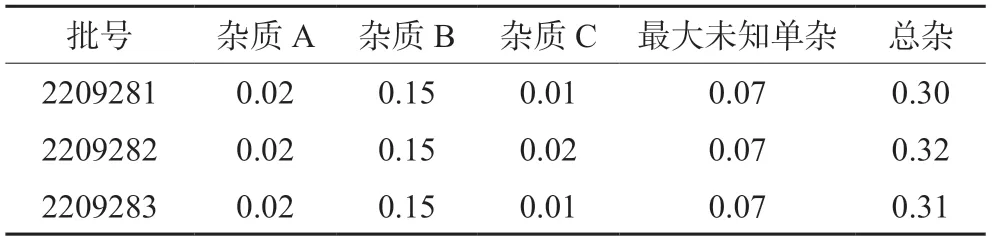
表3 有关物质的含量(%)Tab 3 Content of related substances (%)
3 讨论
用二极管阵列检测器(DAD)对各个物质在200~400 nm内进行扫描,结果表明,主成分及已知杂质的吸收峰均在228 nm及248 nm附近,各已知杂质在248 nm波长下紫外吸收较强且基线平稳。最终选择248 nm作为LPM7100328有关物质的检测波长。
本研究对流动相进行了优化,尝试了磷酸水-乙腈、磷酸缓冲盐-乙腈、磷酸缓冲盐-乙腈-甲醇的流动相体系,筛选并优化了体系中各流动相组分的组成及比例,考察了LPM7100328及其有关物质的分离度、峰形和保留时间,最终确定的流动相A为0.01 mol·L-1磷酸二氢钾水溶液(用磷酸调节pH值至2.5),流动相B为乙腈-甲醇(80∶20)。
为了更好地分离主峰与其相邻杂质峰,本试验选择了Waters XBridge C18(4.6 mm×150 mm,3.5 µm)(Ⅰ)、YMC Triart C18(3 mm×150 mm,3 µm)(Ⅱ)和ACE Excel Super C18(4.6 mm×150 mm,3 µm)(Ⅲ)3根不同型号的色谱柱。相较于色谱柱Ⅰ和Ⅱ,色谱柱Ⅲ具有主峰与其相邻杂质峰的分离情况最佳以及主峰保留时间最稳定的优势,最终选择ACE Excel Super C18(4.6 mm×150 mm,3 µm)为色谱柱,并使用了Welch C18杂质捕集小柱,有效地排除了空白溶剂的干扰,提高了分析结果的准确度[14]。
LPM7100328为正处于Ⅰ期临床研究的1类新药,本研究首次建立了一种LPM7100328原料药中有关物质测定的HPLC分析方法,并对其进行了全面的方法学验证,结果表明该方法专属性好,灵敏度高,可实现LPM7100328中有关物质的有效分离,为LPM7100328质量控制提供参考。
猜你喜欢
云南化工(2021年7期)2021-12-21
中国盐业(2018年20期)2019-01-14
——一个解释欧姆表刻度不均匀的好方法
教学考试(高考物理)(2018年6期)2018-12-06
山东化工(2018年15期)2018-09-20
数学小灵通(1-2年级)(2017年9期)2017-10-13
学苑创造·B版(2017年1期)2017-02-21
小天使·二年级语数英综合(2016年9期)2016-05-14
首都食品与医药(2015年18期)2015-11-03
中华皮肤科杂志(2014年4期)2014-12-19
中华皮肤科杂志(2014年3期)2014-12-19