
丙硫菌唑合成工艺中副产物形成分析
2024-03-11 08:49闫景铭王夏菲程绎南张蒙萌马艺超王利民李洪连
农药学学报 2024年1期
闫景铭, 王夏菲, 程绎南*,,2,3, 张蒙萌, 马艺超, 王利民,2, 李洪连,2
(1.河南农业大学 植物保护学院,郑州 450046;2.河南省植物健康保护技术工程研究中心,郑州 450046;3.河南农业大学 农药纳米工程技术中心,郑州 450046)
丙硫菌唑是一种新型三唑硫酮类杀菌剂,其化学名为2-[(2RS-2-(1-氯代环丙基)-3-(2-氯苯基)-2-羟基丙基]-2H-1,2,4-三唑(4H)硫酮,由德国拜耳公司开发[1],分子结构如图式1 所示。
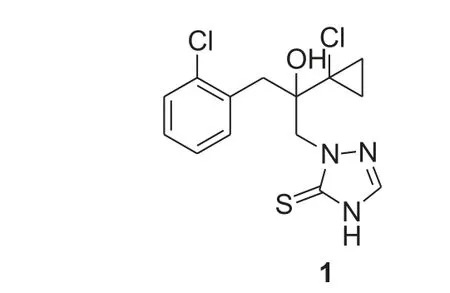
图式 1 丙硫菌唑结构式Scheme 1 The formula of prothioconazole
丙硫菌唑对纹枯病、锈病、白粉病、枯萎病、菌核病、叶斑病、云纹病及网斑病等植物病害均具有较好的防效,广泛应用于麦类作物[2]以及水稻、花生、大豆、油菜等作物的病害防治[3]。目前认为其作用机制是通过抑制细胞色素P450 家族甲基氧化酶CYP51 的活性,进而抑制真菌中羊毛甾醇或2,4-亚甲基二氢羊毛甾醇14 位甲基氧化和脱甲基化作用,阻断甾醇的生物合成[4-5]。丙硫菌唑具有较好的内吸活性、优异的保护活性和治疗活性[6],持效期较长,与其他杀菌剂的交互抗性风险小[7-8],于2019 年在我国登记,主要应用于小麦赤霉病、白粉病、锈病等病害的防治[9]。
由于丙硫菌唑结构的特殊性,其合成及纯化方法面临一定的挑战。目前,按照三唑硫酮结构的构建方式不同,可将丙硫菌唑的合成分为如图式2所示的3 条主要合成路线。
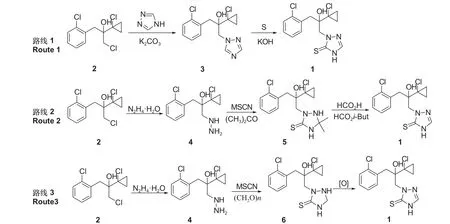
图式 2 丙硫菌唑的3 条主要合成路线Scheme 2 The three main synthetic procedures of prothioconazole
路线1 是以1, 2, 4-三唑为单元构建三唑硫酮结构的合成路线 (三氮唑合成路线)[10],该路线合成步骤少,但在原料1-氯-2-(1-氯环丙基)-3-(2-氯苯基)-2-丙醇(2) 向脱硫丙硫菌唑 (3) 的转化过程中,由于三氮唑不同氮原子参与反应时的选择性不高,导致发生副反应 (图式3 副反应1),使脱硫丙硫菌唑 (3) 的收率仅有50%左右,严重影响该路线工艺收率[11]。
路线2 是以取代肼(4)和丙酮为单元构建三唑硫酮结构的合成路线 (取代肼-丙酮合成路线)[12],该路线所涉及的三唑硫啉酮中间体5 易于分离和纯化,然而,在5 向丙硫菌唑转化时产生的一些未知副产物对产品的分离、纯化以及收率造成了较大负面影响 (图式3 副反应2)。
路线3 是以取代肼(4)和甲醛为单元构建三唑硫酮结构的合成路线 (取代肼-甲醛合成路线)[13],该路线涉及二氢丙硫菌唑(6)的催化氧化及丙硫菌唑的纯化过程。在催化氧化过程中通常伴随丙硫菌唑缩合物的产生 (图式3 副反应3),在纯化过程中如果工艺条件操作不当,也会产生一些副产物 (图式3 副反应4),直接影响产品的质量和收率。
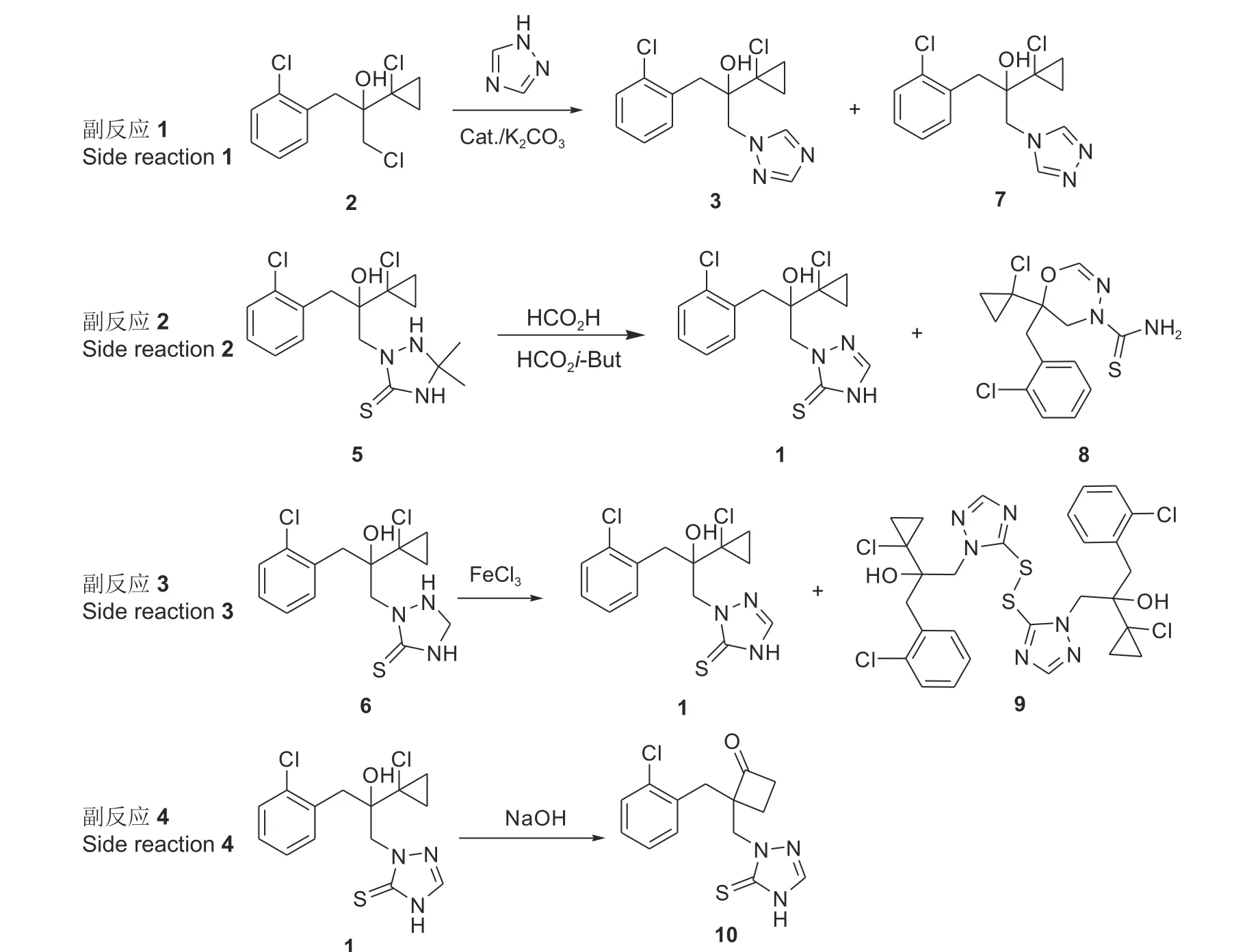
图式 3 丙硫菌唑不同合成路线所涉及的副反应Scheme 3 Side reactions involved in the different synthetic procedures of prothioconazole
不同合成路线发生副反应和后处理过程产生的副产物是影响产品收率和质量的主要因素。为此,本课题组对丙硫菌唑不同合成路线中的关键副产物进行了合成、分离及结构解析,研究其转化特性及其形成机制,旨在为丙硫菌唑的合成、提高与改进其工艺水平提供借鉴与参考。
1 实验部分
1.1 材料与仪器
化学试剂购自上海百舜生物科技有限公司和安耐吉化学有限公司等,所有试剂未经纯化直接使用。薄层色谱板用羧甲基纤维素钠水溶液(3.5‰)与青岛海洋化工厂生产的硅胶 (GF254) 制备。
Bruker DPX-400 型超导核磁共振仪 (德国Bruker 公司),以氘代二甲亚砜 (DMSO-d6) 或氘代氯仿 (CDCl3) 为溶剂,以四甲基硅烷 (TMS) 为内标;安捷伦HP1100 液相色谱-质谱联用仪 (安捷伦科技中国有限公司);SPSIC WRS-1B 数字熔点仪(上海精密科学仪器有限公司); Xcalibur, Eos,Gemini 衍射仪 (安捷伦科技中国有限公司)。除特殊说明,制备色谱均以V(乙酸乙酯) :V(石油醚) =1 : 5 为流动相。
1.2 合成方法
1.2.1 脱硫丙硫菌唑及副产物7 的合成与表征 以正丁醇 (20 mL) 为溶剂,向反应瓶中加入0.83 g(12 mmo1) 1, 2, 4-三氮唑与1.66 g (12 mmo1) 碳酸钾,搅拌混合均匀后,加入2.78 g (0.01 mo1)1-氯-2-(1-氯环丙基)-3-(2-氯苯基)-2-丙醇 (2) 和0.06 g (0.50 mmol) 4-二甲氨基吡啶,加热至回流反应3 h。反应液冷却后加水 (50 mL),用乙酸乙酯萃取(25 mL×3),合并有机相,有机相经水洗后,用无水硫酸钠干燥,抽滤,滤液脱除溶剂得棕色物质3.13 g。粗产物以V(乙酸乙酯) :V(石油醚)= 1 : 20 为流动相进行柱层析分离,得白色固体产物脱硫丙硫菌唑 (3) 1.05 g,收率 34%;同时得另一白色副产物2-(1-氯环丙基)-1-(2-氯苯基)-3-(1H-1,2,4-唑-1-基)-2-丙醇 (7) 0.93 g,收率 30%。脱硫丙硫菌唑3:m.p.107.6~109.4oC;13C NMR(100 MHz, CDCl3)δ: 9.55, 10.80, 37.94, 45.99,53.08, 75.60, 126.62, 128.45, 129.43, 133.57, 133.86,134.84, 145.05, 152.08.1H NMR 与文献[14]一致。副产物7:m.p.119.2~120.8oC;13C NMR (100 MHz,DMSO-d6)δ: 9.23, 10.68, 38.19, 46.06, 49.75, 73.85,126.71, 128.56, 129.26, 133.45, 133.57, 134.21, 144.05.1H NMR 与文献[15]一致。
1.2.2 取代肼-丙酮路线所涉及丙硫菌唑及副产物8 的合成与表征 将64.30 g (0.17 mol) 二甲基三唑硫啉酮 (5) (参照文献[12]合成) 加入到盛有394.80 g(8.58 mol) 甲酸和592 mL 甲酸异丁酯的反应瓶中,加热回流反应16 h,TLC(V(乙酸乙酯) :V(石油醚) = 1 : 5)监测反应物转化完全,停止反应。减压脱除过量的甲酸和甲酸丁酯,残余物溶于400 mL甲苯中,用300.00 g 质量分数为10% 水溶液洗涤,分别得到甲苯相和碱水相。将所得碱水相加入500 mL 甲苯并用10%盐酸酸化到pH 值约为3~4 后分液,有机相加入无水硫酸钠干燥、抽滤脱除溶剂,得到丙硫菌唑粗品52.00 g,甲醇重结晶得到白色针状结晶 (1) 44.10 g,收率74%。甲苯相浓缩,残余物甲醇重结晶得无色固体副产物6-(2-氯苄基)-6-(1-氯环丙基)-5,6-二氢-4H-1,3,4-噁二嗪-4-硫代甲酰胺 (8) 7.70 g,收率13%;副产物8: m.p.154.7~156.3oC;1H NMR (400 MHz,CDCl3)δ: 0.87-1.00 (m, 4H, CH2CH2), 3.45 (d,J=14 Hz, 1 H, CH2), 3.53 (d,J= 14 Hz, 1H, CH2), 4.12(d,J= 14 Hz, 1H, CH2), 4.99 (d,J= 14 Hz,1 H, CH2), 6.24 (s, 1H, NH2), 6.62 (s, 1H, OCHN),7.20-7.22 (m, 3H, ArH and NH2), 7.33-7.35 (m, 1H,ArH), 7.48-7.50 (m, 1H, ArH).13C NMR, (100 MHz,CDCl3)δ: 11.25, 12.26, 36.82, 43.96, 48.15, 79.13,126.40, 128.73, 129.55, 132.19, 133.36, 135.30, 140.27,179.49; HRMS (m/z):C14H16Cl2N3OS [M + H]+,计算值344.0391, 实测值344.0391。
1.2.3 丙硫菌唑缩合物9 的合成与表征 将4.50 g(13.00 mmol)的二氢丙硫菌唑 (6) (参照文献[13]溶于50 mL 甲醇中,加入20% FeCl3(8.40 g) 水溶液,在剧烈搅拌下室温反应5 h。减压浓缩除去溶剂,残液加30 mL 水,用甲苯萃取(20 mL×3),有机相干燥浓缩后,用制备薄层色谱分离得白色固体丙硫菌唑2.70 g,收率60.4% 和丙硫菌唑缩合物(9) 1.40 g,收率31.4%。化合物9:m.p.128.5~130.3oC.1H NMR (400 MHz, CDCl3)δ: 0.43-0.57 (m, 4H, CH2CH2), 0.75-0.78 (m, 2H, CH2CH2),0.95-0.97 (m, 2H, CH2CH2), 3.03-3.08 (m, 2H, CH2),3.64-3.69 (m, 2 H, CH2), 3.91-4.00 (m, 2H, CH2),4.31-4.33 (m, 2H, OH), 5.03-5.08 (m, 2H, CH2), 7.19-7.25 (m, 4H, ArH), 7.37-7.39 (m, 2H, ArH), 7.51-7.53 (m, 2H,ArH), 7.96(s, 1H, Triazole-H), 7.98(s, 1H, Triazole-H);13C NMR, (100 MHz, CDCl3)δ: 10.09, 10.17,10.95, 11.04, 38.12, 38.16, 45.60, 53.13, 53.24, 75.68,126.60, 128.47, 129.42, 133.54, 133.58, 133.81, 133.84,134.89, 151.73, 151.86, 152.24.HRMS (m/z):C28H29Cl4N6O2S2[M + H]+, 计算值687.0548, 实测值687.0543.熔点与文献(m.p.128.0~131.0oC)[16-17]一致。
1.2.4 碱处理副产物10 的合成与表征 将丙硫菌唑 (1)(5.00 g) 溶于10% NaOH (1.20 g)水溶液中,室温搅拌反应10 h。反应液用30 mL 甲苯萃取得到有机相,有机相经水洗、分离、干燥后浓缩。残余物用薄层色谱分离得0.20 g 副产物2-(2-氯苄基)-2-((5-硫酮-4,5-二氢-1,2,4-三唑-1-基)甲基)环丁酮 (10),白色固体,收率4.5%。副产物10:m.p.101.9~103.8oC.1H NMR (400 MHz, CDCl3)δ: 1.96-2.04(m, 1H, CH2CH2), 2.17-2.25 (m, 1H, CH2CH2), 2.58-2.67 (m, 1H, CH2CH2), 2.90-2.99 (m, 1H, CH2CH2),3.19 (d,J= 14 Hz, 1 H, CH2), 3.28 (d,J= 14 Hz, 1H,CH2CH2), 4.39 (d,J= 14 Hz, 1H, CH2CH2), 4.63 (d,J= 14 Hz, 1H, CH2CH2), 7.19-7.22 (m, 2H, ArH),7.27-7.30 (m, 1H, ArH), 7.38-7.40 (m, 1H, ArH),7.89 (s, 1H, Triazole-H), 12.74 (brs, 1H, NH).13C NMR (100 MHz, CDCl3)δ: 19.10, 35.34, 44.41, 50.76,68.65, 127.05, 128.48, 129.81, 132.04, 134.22, 134.98,137.82, 166.11, 210.94.HRMS (m/z):C14H15ClN3OS[M + H]+,计算值308.0624, 实测值308.0623.熔点与文献(m.p.102.5~103.5oC)[18]一致。
1.2.5 丙硫菌唑与丙硫菌唑缩合物9 的转化 称取丙硫菌唑0.34 g (1.00 mmol),投入盛有25 mL甲醇的三口烧瓶中,加入20% FeCl3(1.62 g)水溶液,在50oC (甲醇微负压脱溶温度) 剧烈搅拌下反应8 h,真空浓缩。残液加30 mL 水,用甲苯萃取(20 mL×3),合并有机相并干燥浓缩后,以乙酸乙酯和石油醚 (V:V= 1 : 10) 为展开剂,用薄层色谱分离得0.23 g 淡黄色固体丙硫菌唑缩合物9,收率67%。
称取0.69 g (1.00 mmol) 化合物9 投入50 mL 三口烧瓶中,加入四丁基溴化胺3.25 mg (0.01 mmol)、连二硫酸钠(Na2S2O4) 0.44 g ( 2.50 mmol) 和水20 mL,搅拌反应4 h。反应液用乙酸乙酯萃取(2 0 mL×3),合并有机相并干燥浓缩后,残余物以V(乙酸乙酯) :V(石油醚) = 1 : 10 为展开剂,用薄层色谱分离得白色固体丙硫菌唑0.56 g,收率81%。
2 结果与讨论
2.1 丙硫菌唑三氮唑合成路线主要副产物结构及产生机制分析
在丙硫菌唑三氮唑合成路线中,首先通过1,2,4-三唑中的氮原子向原料2 中的碳-氯键发生亲核取代反应引入三唑结构,然后再通过硫的氧化反应合成丙硫菌唑[11]。但在该合成步骤中,无论反应条件如何优化,总有一个副产物产生,且与目标产物数量相当,严重影响了目标产物的收率和中间体的纯化。高分辨质谱显示,其分子质量与脱硫丙硫菌唑 3 相同。核磁图谱显示,三唑官能团的氢信号在δ8.37 的低场处显示1 单峰为2 个氢,与脱硫丙硫菌唑 3 分别在δ8.26 和δ7.95处出现1 单峰各1 个氢形成鲜明对比,出现了氢信号的简并现象;同时其碳信号在δ144.05 的低场处也出现了简并,说明三唑的连接方式具有对称性特征,因此判断其为如图式3 所示结构的副产物7。分析其形成机制为在碱性条件下,1,2,4-三唑被夺去质子后,存在两种共振氮负离子(如图式4 所示),从而导致副产物7 的产生。改变实验条件,如降低反应温度,改变三氮唑的投料比以及缚酸剂等,该副产物相对于目标产物的比例均没有明显改变。目前,副产物7 并不能通过反应条件的优化得到明显改善,这是制约该合成路线的严重不利因素。

图式 4 副产物7 可能的形成机制Scheme 4 The possible mechanism of formation of by-product 7
2.2 取代肼-丙酮合成路线主要副产物结构及产生机制分析
在取代肼-丙酮的合成路线中,涉及如图式2 路线2 所示的合成过程,其中中间体5 通过取代肼、硫氰酸盐和丙酮的缩合而成[12]。由于丙酮参与的硫酮杂环构建,反应的区域选择性强,所产生的中间体5 容易分离,该合成路线具有一定的优势。然而,中间体5 在甲酸及甲酸酯的存在下向丙硫菌唑转化过程中,伴随一个主要副产物的产生。从核磁图谱分析,其氢及碳信号与丙硫菌唑在数量上完全一致,质谱分析表明其分子质量也与丙硫菌唑完全相同,因此难以确定其结构。为此,培养了副产物8 的晶体进行了X-射线衍射分析,其晶体结构如图1 所示 (CCDC 号为2305552),最终确定其为具有如图式3 化合物8 所示结构的化合物。
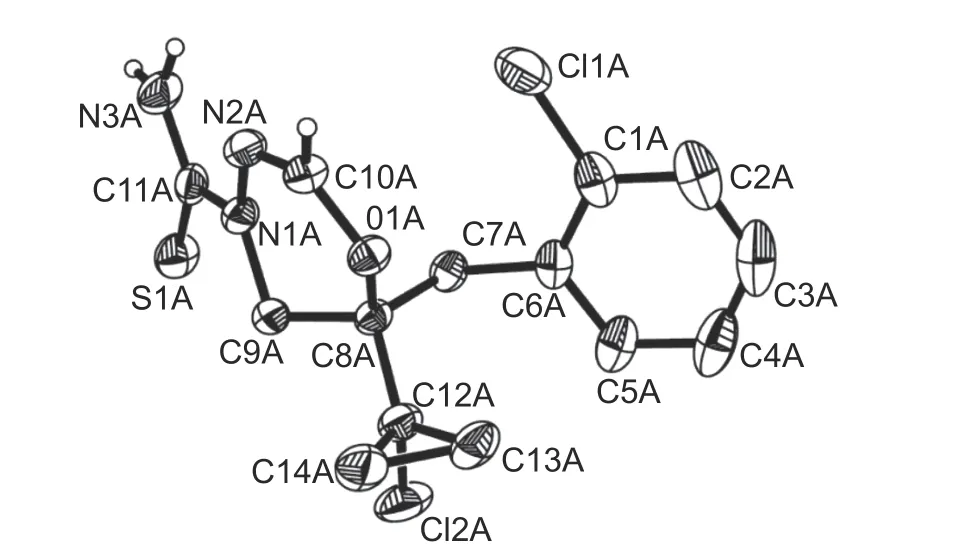
图1 副产物8 的晶体结构Fig.1 Crystal structure of by-product 8
化合物8 的形成出乎意料,原料分子骨架中的叔羟基参与了杂环的重组过程。可能的形成机制如图式5 所示,首先,三唑硫啉酮(5) 1 位的氮原子与甲酸酯发生酯胺交换反应;其次,三唑硫啉酮在水的存在下水解开环;接着,在酸的催化下,羟基向羰基发生加成、消除反应,形成噁二嗪环;最后取代硫脲的氮原子上进一步发生水解反应,释放出一分子丙酮和水,最终形成副产物8。在目前实验条件下,副产物8 相对于中间体5约有13%的收率 (见实验部分),严重影响丙硫菌唑的工艺收率、产品纯化和质量。

图式 5 副产物8 可能的形成机制Scheme 5 The possible mechanism of formation of by-product 8
2.3 取代肼-甲醛合成路线副产物结构及产生机制分析
在取代肼-甲醛的合成路线中,取代肼在硫氰酸盐和甲醛的存在下缩合得到二氢丙硫菌唑 (6)(图式2 路线3)。6 在氧化剂的存在下氧化脱氢,转化为丙硫菌唑 (1),目前多以水-醇溶液为媒介,采用三氯化铁为氧化剂实现氧化过程 (图式6)。然而,在氧化步骤中产品收率波动较大。分析其原因发现,在氧化过程中,不同操作条件下除生成丙硫菌唑以外,还伴随一个主要副产物的生成,从而造成收率的降低。1H NMR 图谱表明,该副产物氢信号与丙硫菌唑相比少了一组活泼氢(酰胺N-H),其他出峰位置及裂分与丙硫菌唑相似。HPLCMS 显示,其质荷比[M + H]+/z为687.0543,表明其相对分子质量为丙硫菌唑的2 倍少两个质量数,判断该产物为丙硫菌唑缩合物,其结构如图式3化合物9 所示。该化合物的熔点和1H NMR 与文献[16-17]基本一致,所不同的是看似对称的质子及碳信号并不能完全重叠。丙硫菌唑分子中叔醇碳原子为手性碳,丙硫菌唑是(R/S)构型消旋体,而当两分子间发生缩合时会生成(R/S)、(S/R)、(R/R)和(S/S)4 个异构体,其中(R/S)和(S/R)的NMR 信号完全相同,(R/R)和(S/S) 的NMR 信号也完全相同。但(R/S)和(S/R)与(R/R)和(S/S) 是差向异构体,它们的NMR 信号十分相似并不完全相同,如1H NMR 谱中在δ7.98和7.96 出现两组三唑环的质子单峰,13C NMR谱在δ10.09 和10.17,10.95 和11.04, 38.12 和38.16 以及53.13 和53.24等处出现成对的共振信号,证明了这种差向异构体的存在。但这一细微差别只有在较高分辨率的情况下才能观察到。
至于该副产物的形成机制,由于丙硫菌唑具有如图式6 所示硫酮式1 和硫醇式11 两种互变异构体,而硫醇通常在氧化剂或酸的存在下发生缩合反应[19],因此判断缩合物9 由丙硫菌唑硫醇式11 氧化缩合生成 (图式6)。为了进一步证实这一转化,直接将分离纯的丙硫菌唑在二氢丙硫菌唑相似的氧化条件下使用三氯化铁处理,最后以67%的收率分离得到了缩合体9。

图式 6 丙硫菌唑缩合物9 可能的形成机制Scheme 6 Possible mechanism of formation of dimer 9
进一步研究发现,缩合体9可以在 Na2S2O4存在下解聚[20],转化为丙硫菌唑 (图式6)。为此,将丙硫菌唑缩合物在相转移催化剂存在下用保险粉水溶液处理,以81%的收率将丙硫菌唑缩合物还原为丙硫菌唑 ,此方法可以解决丙硫菌唑因缩合造成的收率损失问题。
2.4 丙硫菌唑纯化过程中副产物结构及产生机制分析
由于丙硫菌唑具有溶于碱性水溶液的特点,通常使用氢氧化钠水溶液将其溶解,先萃取去除有机杂质,再酸化水相得到丙硫菌唑并纯化。然而在纯化过程中,通常伴随一个少量杂质的产生。
核磁图谱显示,该杂质的Ar-CH2以及三唑N-CH2信号清晰可见,而相对高场质子信号发生了较大变化,其化学位移较丙硫菌唑向低场移动,4 个质子都分别裂分为多重峰 (δ1.96~2.99),表明它们之间彼此仍是互相靠近的,羟基质子信号消失,表明环丙烷及其邻近结构发生了变化。结合碳谱在化学位移为δ210.94 处的低场出现一个碳信号,说明存在1 个羰基碳。HPLC-MS 显示,其质荷比[M + H]+/z为308.0623,与理论值308.0624对应。结合以上特征,综合判断其为图式3化合物10所示的结构,参考相关文献[18]进一步印证了结构的正确性。
在文献[18, 21]中,虽然提及该化合物为丙硫菌唑杂质,但并没有分析其形成原因。我们分析该化合物的形成可能经历了如图式7所示的步骤:首先,丙硫菌唑在碱的存在下在环丙烷端发生C-Cl 键的水解或羟基向C-Cl 键的亲核取代,分别形成邻二醇12或双环结构13[22],然后再分别发生重排反应[23]生成副产物10。因此,在丙硫菌唑纯化处理过程中,控制氢氧化钠的浓度、处理温度、与氢氧化钠的接触时间以及后续的酸化条件等,是防止该副产物产生的关键因素。
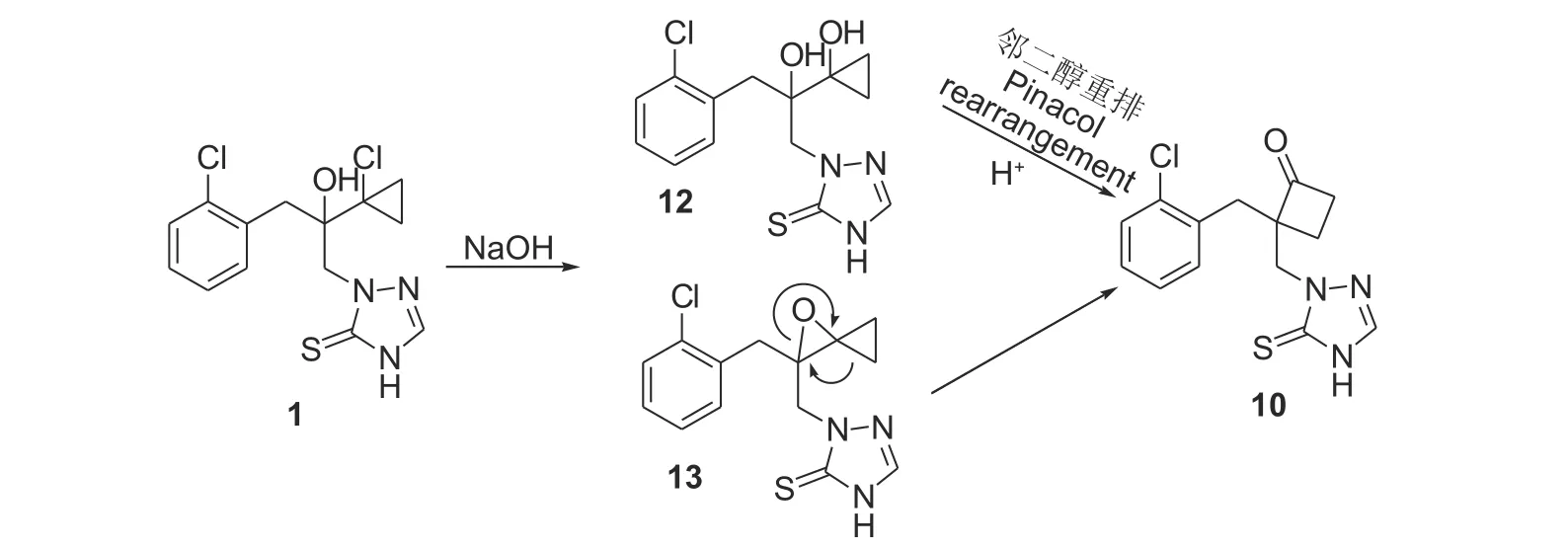
图式 7 丙硫菌唑碱及酸处理副产物10 的可能形成机制Scheme 7 Possible mechanism of formation of by-product 10 in prothioconazole treatment with base and acid in sequence
3 结论
通过对丙硫菌唑不同合成路线中副产物的分离,借助1H NMR、13C NMR、ESI-MS 或 X-射线衍射等手段对其结构进行了表征,分别明确了三氮唑合成路线、取代肼-丙酮合成路线、取代肼-甲醛合成路线以及丙硫菌唑纯化工艺中的主要副产物结构,并分析了其形成机制。提出了丙硫菌唑缩合物形成及控制措施,丙硫菌唑纯化中碱处理需要注意的关键因素等。丙硫菌唑不同合成路线主要副产物的表征、形成机制分析以及相关控制措施的验证,为丙硫菌唑产品的合成路线选择、工艺控制及产品质量的提高提供了重要参考。
猜你喜欢
安徽农业大学学报(2022年2期)2022-11-09
保鲜与加工(2021年1期)2021-02-06
农药科学与管理(2019年7期)2019-11-29
农药科学与管理(2019年10期)2019-04-20
广东饲料(2016年5期)2016-12-01
今日农药(2016年8期)2016-11-15
中国果菜(2016年9期)2016-03-01
中国资源综合利用(2016年12期)2016-01-22
西华师范大学学报(自然科学版)(2015年3期)2015-02-27
应用化工(2014年12期)2014-08-16
