
催化剂中金属与载体相互作用的表征研究进展
2024-03-11 08:24彭洁李曜储政于杨
天然气化工—C1化学与化工 2024年2期
彭洁,李曜,储政,于杨
(中石化南京化工研究院有限公司,江苏 南京 210048)
早在1978年,相关人员在研究TiO2负载的贵金属催化剂时,提出了强金属-载体相互作用(SMSⅠ)的概念[6-7]。之后,MSⅠ得到了更广泛关注。MSⅠ普遍存在于负载型催化剂中,有多种表现形式,包括传统经典的SMSⅠ、氧化金属-载体相互作用(OMSⅠ)、共价金属载体相互作用(CMSⅠ)和电子金属-载体相互作用(EMSⅠ)等[8-13],这些相互作用的具体表现形式由活性金属和载体的类型和性质决定。MSⅠ通常会导致载体对金属的“包覆”、界面电荷转移和催化剂对小分子的吸附减弱等现象,进而影响催化剂的催化活性、选择性及稳定性[11,13]。
近年来,随着表征技术的不断发展,特别是原位表征方法和高分辨电镜技术的进步,人们对负载型催化剂的认识也不断深入,从表观的结构特征到金属-载体界面原子间的电荷转移,从静态物化性质到实际反应过程中的相互作用动态变化,MSⅠ的表征研究已非常广泛[11,14]。但是目前对于MSⅠ的报道多是对某一种或某几种催化剂材料进行针对性研究,相关综述文章多是从MSⅠ的概念、形成、影响或应用展开,虽然部分文章会列举使用到的表征方法,但对MSⅠ的表征方法的系统性总结较少[10,14-15]。本文基于非原位和原位表征技术,包括高分辨透射电子显微镜(HRTEM)、拉曼光谱(Raman)、X射线光电子能谱(XPS)和电子顺磁共振谱(EPR)等,从MSⅠ的静态性质(如物质输送导致的微观结构特征、电荷转移等)和动态性质(相互作用强弱变化即作用可逆性、界面结构变化和电荷转移过程等)展开,概述MSⅠ的表征研究进展(图1)。
1 MSⅠ的非原位表征
通过非原位手段,可以表征催化剂预处理后或反应前后稳定状态下的MSⅠ。例如,HRTEM可以观测金属-载体界面的微观结构,从而得到质量转移信息;红外光谱(FTⅠR)、Raman等可以表征金属-载体之间的成键情况;XPS、EPR等可以用于分析金属-载体之间的界面电荷转移。
1.1 MSI下的界面微观结构表征
SⅠNGH等[16]早在1985年便通过HRTEM观测到了Rh/TiO2催化剂中Rh颗粒表面包覆的氧化物载体,这种“包覆”表明Rh与TiO2载体之间存在强相互作用,之后HRTEM逐渐成为表征金属-载体界面结构和质量转移的最常用方法[17-20]。早期人们认为由于Au的功函和表面能较低,不易产生SMSⅠ,但近年来,对于Au-载体之间相互作用的研究逐渐增多。经过高温焙烧之后,Au与载体之间可以产生不同程度的相互作用。TANG等[19]研究发现,羟磷灰石(HAP)载体在Au纳米颗粒表面的包裹程度取决于焙烧温度(图2)。由图2可知,焙烧温度为300 ℃时,纳米颗粒开始被薄层覆盖,随着温度升高,覆盖程度逐渐增大,600 ℃时Au纳米颗粒被完全覆盖。之后,Au/H-500-H2样品(数字为焙烧温度)在H2氛围下还原,覆盖层逐渐褪去。为了确认覆盖层的组分,还对Au/H-600样品进行了电子能量损失谱(EELS)分析,结果显示覆盖层信号与HAP载体相同,即表明在焙烧过程中载体迁移至Au纳米颗粒表面,形成SMSⅠ。类似地,LⅠU等[20]通过HRTEM观测到Au/ZnO催化剂经过300 ℃氧化处理之后,ZnO纳米棒载体组分向Au纳米球迁移,包覆层的晶格间距为0.26 nm,与ZnO(002)晶面一致。H2氛围下还原后,包覆层褪去,符合SMSⅠ的典型特征。
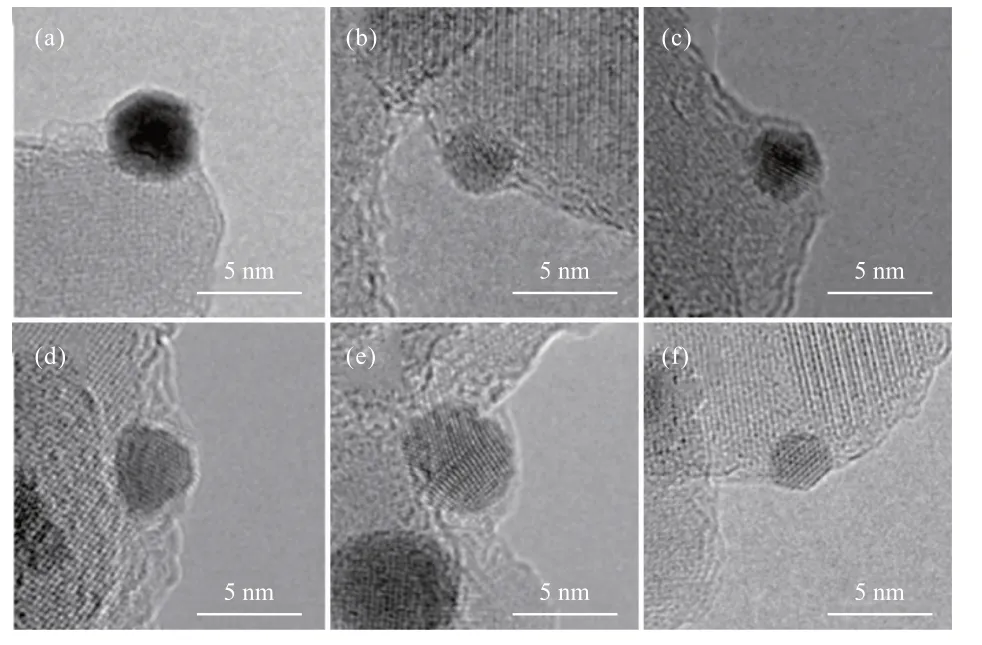
图2 Au/H-200 (a)、Au/H-300 (b)、Au/H-400 (c)、Au/H-500 (d)、Au/H-600 (e)和Au/H-500-H2 (f)的HRTEM照片[19]Fig.2 HRTEM images of Au/H-200 (a), Au/H-300 (b), Au/H-400 (c),Au/H-500 (d), Au/H-600 (e) and Au/H-500-H2 (f)[19]
除HRTEM之外,其他高分辨电子显微镜[21-22],如高角环形暗场像-扫描透射电子显微镜(HAADFSTEM)也可用于观测载体与金属纳米颗粒之间的包覆情况。同时,通过漫反射红外光谱(DRⅠFT)探测催化剂对探针分子(如CO、H2)的吸附行为,亦可以侧面说明金属与载体之间的包覆现象,而原位DRⅠFT更是常用于探测这种包覆的变化,进而说明相互作用的强弱变化,这部分内容将在2.1小节举例阐述。
1.2 金属-载体的化学键表征
很多时候,金属-载体之间的相互作用可以用化学键来描述,特别是对于SMSⅠ[23]。事实上,形成化学键是电荷转移的一种表现结果。例如当一个电子从金属转移至载体(如金属氧化物)上时,二者之间就可能形成金属—氧键;而如果金属-载体之间的相互作用较弱,则可能只有部分电荷偏移。上述两种情况均普遍存在于MSⅠ的体系中。
X射线吸收精细结构(XAFS)谱图包括X射线吸收近边结构(XANES)谱图和扩展X射线吸收精细结构(EXAFS)谱图两部分,其具有元素分辨性,可以提供凝聚态物质的结构信息[24-25]。通过XANES谱图可以获得材料的对称性和化学态信息;通过EXAFS谱图可以获得吸收原子的配位环境参数,如原子种类、配位数和原子间距等。因此,XAFS经常被用于表征金属-载体的界面相互作用,确定金属与氧化物载体(如CeO2)之间的键合特征[26-28]。例如,2019年,KOTTWⅠTZ等[28]研究了单原子分散的Pt与CeO2载体之间的相互作用,通过XAFS谱图结合计算模拟,证实了Pt—O—Ce键的形成。CeO2负载单原子Pt材料(Pt SAC/CeO2)的XANES谱图与在k空间和R空间的EXAFS谱图见图3,同时为了对比,图3提供了Pt金属箔、α-PtO2和负载型Pt纳米颗粒(Pt NP/CeO2)的数据[28]。由图3可知,纳米Pt材料与块体Pt材料的EXAFS谱图的特征峰完全不同。进一步分析数据并进行拟合可知,在Pt SAC/CeO2中,与Pt紧邻的是4个O,Pt与O之间的距离为1.995 × 10-10m,同时,Ce与Pt的距离为3.340 × 10-10m,这分别与密度泛函理论(DFT)优化后的PtCe40O80纳米结构模型中Pt—O键长和Pt与Ce的间距接近,证明Pt SAC/CeO2中形成了Pt—O—Ce键。
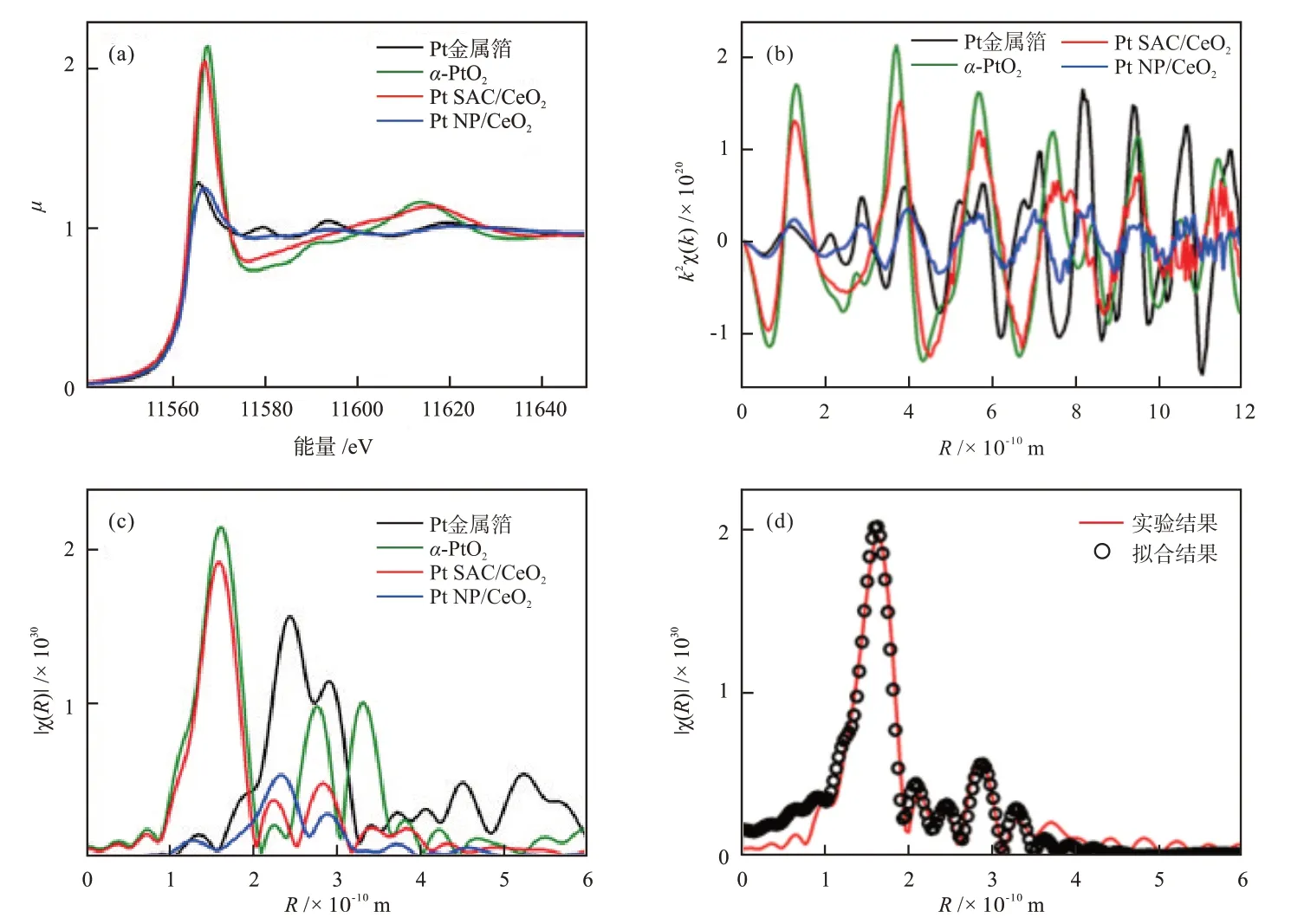
图3 不同Pt材料的XANES谱图(a)、k空间(b)和R空间(c)的EXAFS谱图及Pt SAC/CeO2的实验与拟合结果(d)[28]Fig.3 XANES spectra (a), EXAFS spectra in k space (b) and R space (c) of different Pt materials and experimental and fitting results of Pt SAC/CeO2 (d)[28]
此外,振动和转动光谱用于表征化学键更加直观。从原理上讲,FTⅠR和Raman均可用于表征金属-载体之间的成键情况,如金属—氧键、金属—金属键等[23,29],二者原理不同但信息相似,在使用过程中存在一定的互补性。与FTⅠR相比,Raman可以提供低波数(低于400 cm-1)的振动信息、更适用于无机材料的检测,而FTⅠR的探测范围通常是400~4000 cm-1,非对称分子的红外信号比Raman信号更强。LEE等[29]通过Raman分析证明,当Pt浸渍于CeO2载体之后,Pt通过Pt—O—Ce键锚定在了载体表面,即产生了SMSⅠ。由图4可知,随着Pt含量的增加,属于Pt—O—Ce键的新振动峰,即567 cm-1(Pt—O—Ce键)和655 cm-1(Pt—O键)处振动峰的振动强度逐渐增大。BUKHARⅠ等[30]使用FTⅠR研究了纤维型SBA-15负载的Ni催化剂的MSⅠ,发现载体SBA-15中Si—OH键振动强度的减弱侧面证明了Si—O—Ni键的形成。
除此之外,在需要做好急流动性参数控制,在正式操作过程中,压浆泵需要始终保持开启状态,并在压浆泵中注入含量适中的水分,不断搅拌,促使浆液最终到达最佳浓度和黏稠状态。当浆液黏稠度达到合适状态后,需要用亚水泵将浆液直接输送到孔洞中进行施工。施工过程需要不断保证持续性,施工顺序需要按照从上至下一次完成,除此以外,还需要关注浆体时间。浆液关闭时需要同时关闭压浆泵,如果没有关闭,则需要提高0.5MPa压力,确保压浆泵可以在自然条件下自动停止工作。
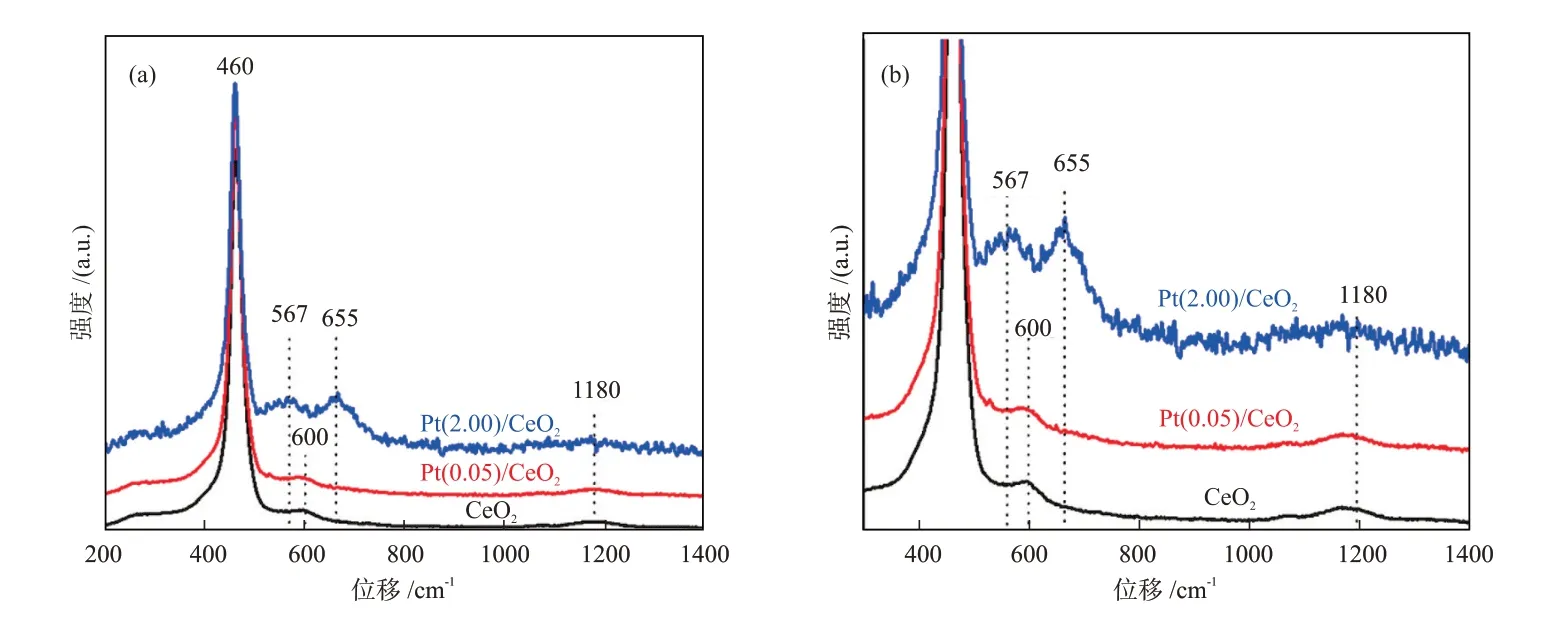
图4 CeO2、Pt(0.05)/CeO2和Pt(2.00)/CeO2的Raman谱图(a)和300~1400 cm-1局部放大谱图(b)[29]。Fig.4 Raman spectra (a) and locally amplified spectra in 300~1400 cm-1 (b) of CeO2, Pt(0.05)/CeO2 and Pt(2.00)/CeO2[29]
1.3 金属-载体的电荷转移表征
一般地,MSⅠ下的电荷转移表现为表面元素价态和电子结构的变化,因此可以使用元素敏感的表征技术进行检测,如XPS、紫外光电子能谱(UPS)、俄歇谱(AES)等[31-35]。例如,LU等[36]研究了一系列贵金属纳米催化剂(Pt、Pd、Rh、Ⅰr和Ag)与TiO2载体之间的相互作用,部分研究结果见图5。由图5可知,对于Pt/TiO2,电子结合能位于71.0 eV、71.9 eV和72.9 eV处的XPS峰分别归属于Pt0、Pt2+和Pt4+,而Pt0的XPS峰一般出现在71.5 eV,即在该催化剂中,Pt0的XPS峰向低能量偏移,说明Pt带负电荷。Pd/TiO2也表现出类似的XPS峰偏移,表明Pt/TiO2和Pd/TiO2通过MSⅠ诱导TiO2局部电荷转移到贵金属纳米颗粒上,这种电荷转移使Pt/TiO2和Pd/TiO2比另几种贵金属催化剂表现出更好的催化性能。
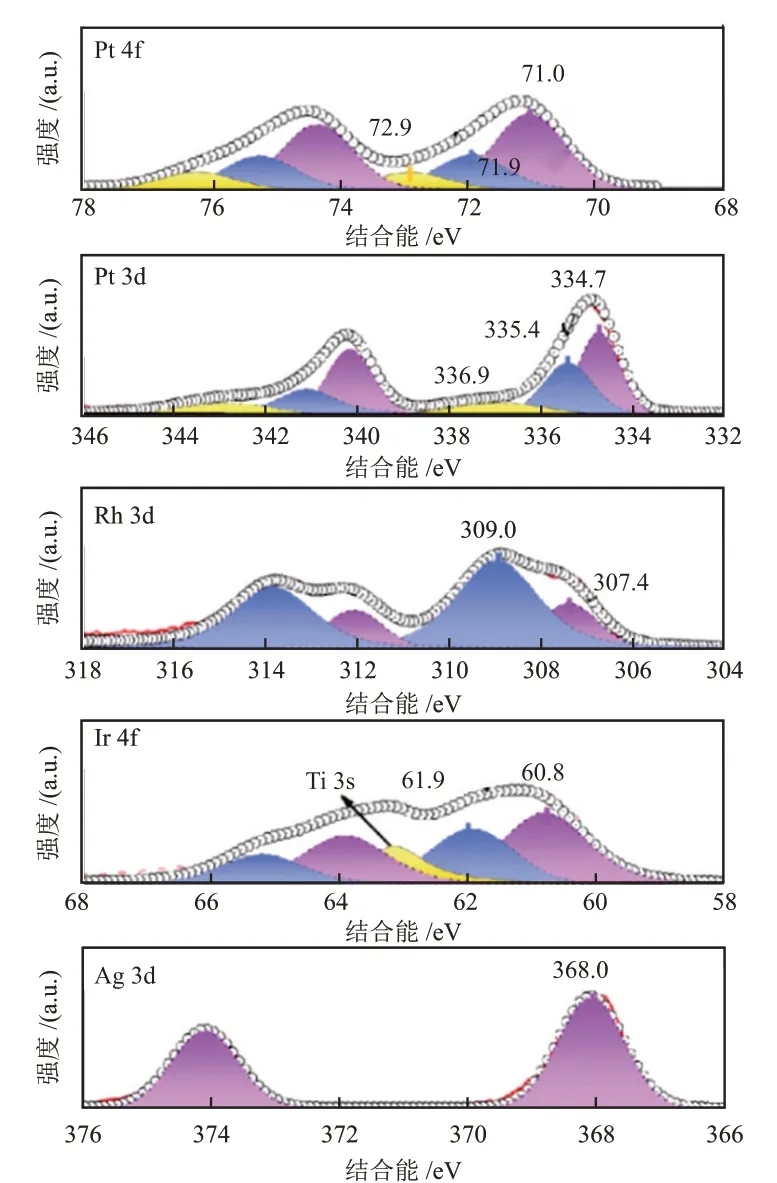
图5 M/TiO2的XPS谱图[36]Fig.5 XPS spectra of M/TiO2[36]
此外,电荷转移必然会改变载体的局部配位环境,氧化物载体的电子结构扰动可通过EPR进行表征[37-39]。WANG等[40]采集了ZnO和Ni/ZnO样品的EPR谱图并探究了Ni和ZnO之间电荷转移路径以及SMSⅠ作用对CO甲烷化反应的影响(图6)。ZnO晶格中存在VOo、VO˙和VO˙ 3种氧空位,其中只有VO˙含未成对电子,具有EPR活性。由图6可知,随着还原温度的升高,与ZnO的EPR信号(归属于VO˙)相比,Ni/ZnO的信号逐渐减弱,说明ZnO载体的VO˙失去电子或得到电子,形成了没有EPR活性的VOo或VO˙。通过XPS谱进一步确认可知,Ni和ZnO粒子之间最可能的电子转移路径是ZnO晶格中的VO˙将电子传递给Ni,形成VOo。
用DRⅠFT表征催化剂对探针分子(如CO)的吸附行为,也可反映催化剂表面的电荷分布情况,例如,CO—Au键的红外吸收带从2101 cm-1蓝移至2113 cm-1,表明Au纳米颗粒表面正电荷增加[20]。
以上主要从MSⅠ的静态性质展开,分别介绍了金属-载体界面微观结构、二者之间成键和电荷转移情况的常用非原位表征方法,其总结见表1。
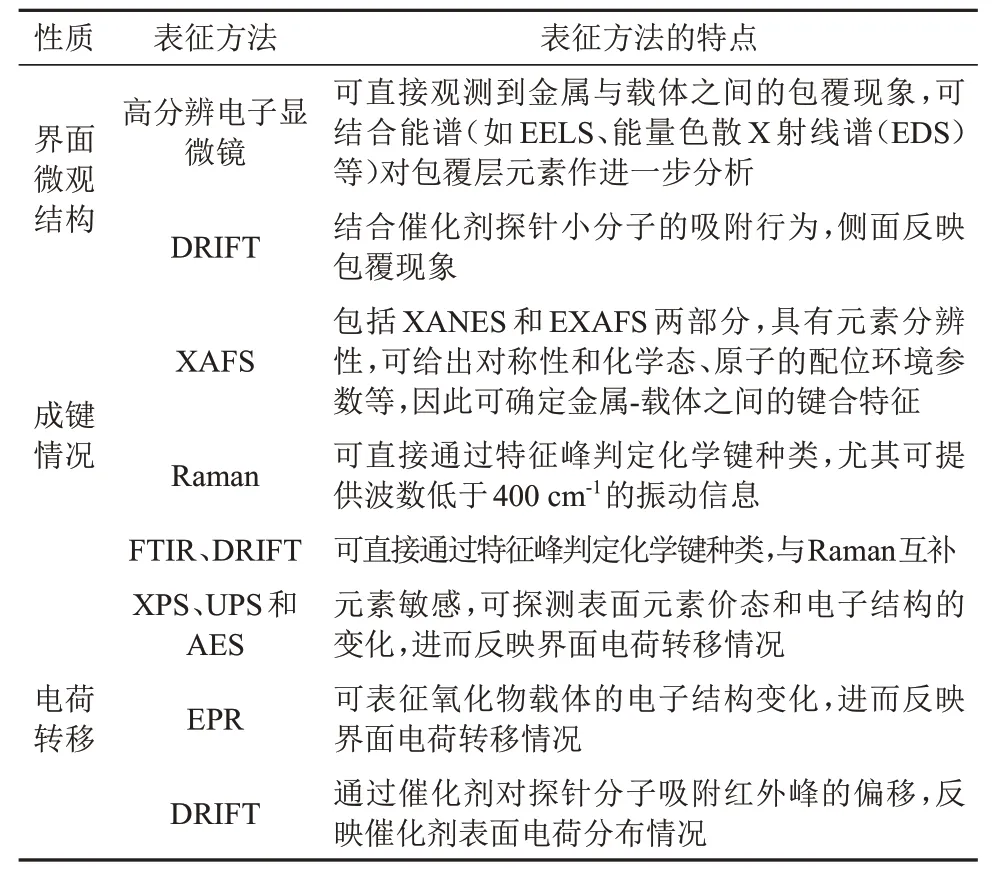
表1 MSI的非原位表征方法总结Table 1 Summary of ex situ characterization methods for MSI
2 MSⅠ的原位表征
MSⅠ具有可逆性,催化剂预处理条件和反应条件均可能使相互作用发生变化。因此,在预处理过程和反应过程中对MSⅠ进行原位动态表征十分必要。一方面有利于理解相互作用的产生机制,另一方面有利于深入探究相互作用对于催化活性和催化反应机制的影响。基于原位表征技术,人们对MSⅠ的强弱变化、反应过程中的界面结构和组分变化,以及电荷转移的动态过程展开了研究。
2.1 MSI的强弱变化表征
催化剂的MSⅠ增强会导致其对小分子的吸附减弱。因此当选择合适的探针分子时,原位DRⅠFTS可以敏锐地探测出金属表面的吸附性能,是表征MSⅠ强弱变化最常用的手段[31,41-43]。TANG等[44]分别研究了Au纳米颗粒与TiO2(Au/T)、HAP(Au/H)和TiO2/HAP(Au/TH)载体之间的相互作用情况。由图7(a)可知,相比于未煅烧的样品(Au/载体-uc),800 ℃煅烧之后的3种Au催化剂(Au/载体-800)的CO振动峰均大幅减弱,说明此时载体对Au纳米颗粒形成包覆,Au与载体TiO2和HAP之间均存在强相互作用。进一步地,在温度为-130 ℃时,对预吸附了CO的Au/TH-800样品通入O2,采集了30 min内的原位DRⅠFT谱图(图7(b))。由图7(b)可知,吸附于TiO2上的CO峰(CO/TiO2)的强度减弱,CO/Au峰的强度不变,CO2振动峰的强度增大,说明载体中引入的TiO2有利于CO催化氧化反应。
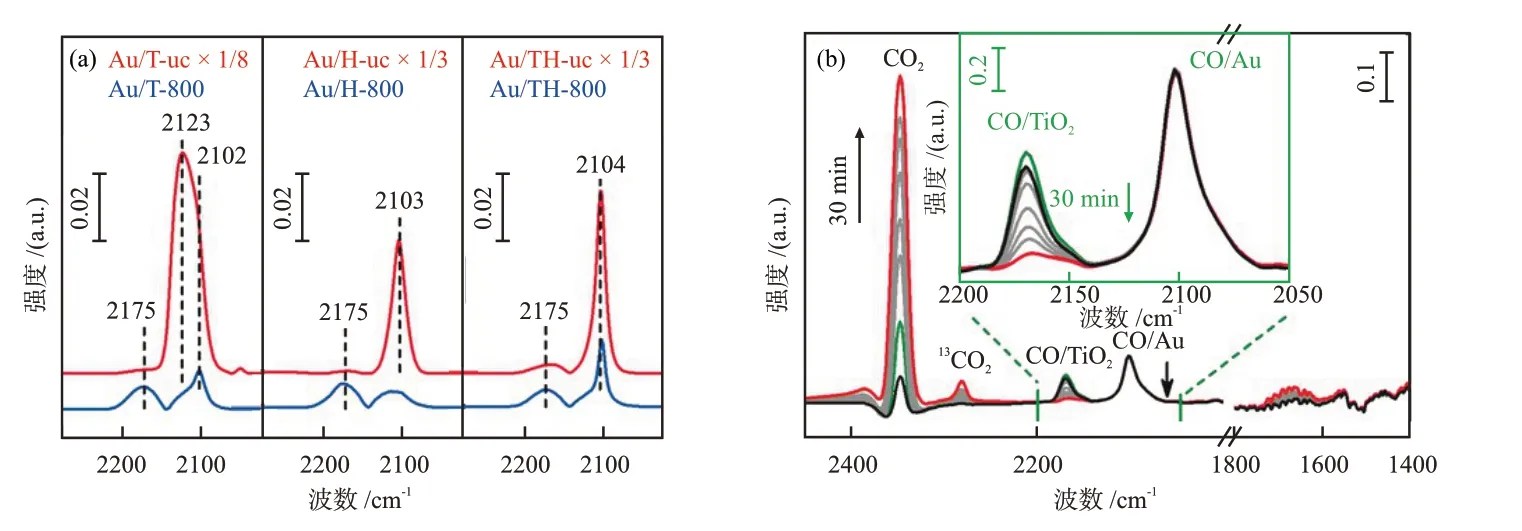
图7 吸附CO 10 min后不同Au催化剂的原位DRIFT谱图(室温)(a)和在预吸附了CO的Au/TH-800中通入2.0 kPa O2后Au/TH-800的原位DRIFT谱图(-130 ℃)(b)[44]Fig.7 In situ DRIFT spectra of different Au catalysts after 10 min of CO adsorption (room temperature) (a) and in situ DRIFT spectra of Au/TH-800 pre adsorbed with CO after introducing 2.0 kPa O2 (-130 ℃) (b)[44]
此外,H2程序升温还原(H2-TPR)也是一种常见的表征相互作用强弱的手段。例如,铜基催化剂的H2还原温度较纯CuO相比有一定幅度的下降,由此可说明存在MSⅠ。CAO等[45]通过H2-TPR研究了负载CeO2、ZrO2、CeO2-ZrO2的Cu催化剂,发现CuCeZr、CuCe和CuZr催化剂的H2还原峰分别位于142 °C、154 °C和162 °C,表明3种催化剂中Cu与载体的相互作用依次减弱。当然,H2-TPR是一种非原位方法,这里仅作为补充方法进行说明,H2-TPR只能表征催化剂的相互作用强弱,而不像原位DRⅠFTS可以表征强弱变化,但H2-TPR是催化剂的常用表征手段,比原位DRⅠFTS更为经济。
2.2 金属-载体之间的界面结构变化表征
基于原位电镜、原位X射线谱学等手段,可以实时观测催化剂的结构演变[46-52]。DONG等[49]发现在甲烷干重整(DRM)反应中,弱氧化性气体可诱导Ni纳米颗粒与六方氮化硼(h-BN)纳米片之间的相互作用,这种相互作用来源于气体刻蚀h-BN载体产生的超薄氧化硼(BOx)覆盖层对Ni颗粒的包覆。原位环境扫描透射电镜(ESTEM)展现了CO2(约3.5 Pa)对h-BN的原位刻蚀过程,通过原位场发射扫描电子显微镜(FESEM)对比了800 ℃时不同气体(H2、CO2、H2O和O2)对MSⅠ的影响,发现弱氧化性气体(CO2、H2O)可以促进Ni与h-BN的界面反应(图8)[49]。

图8 100 ℃ (a)和676 ℃ (b)下Ni/h-BN在CO2中的原位ESTEM照片及800 ℃下Ni/h-BN在H2、CO2、H2O 和O2中的原位FESEM照片(c)[49]Fig.8 In situ ESTEM images of Ni/h-BN in CO2 at 100 ℃ (a)and 676 ℃ (b) and in situ ESTEM images of Ni/h-BN in H2, CO2, H2O and O2 at 800 ℃ (c)[49]
BECK等[48]基于原位电镜、原位XPS和原位XRD,辅以DFT计算,研究了Pt与TiO2载体相互作用的形成过程和结构变化机理,揭示了H2和O2在该过程中的作用。先在600 ℃、100 kPa H2氛围中对样品进行还原,然后在O2氛围下处理,之后再次转换为H2氛围。对上述处理过程进行原位表征可知,H2氛围下,TiO2迁移形成覆盖层和形成Pt-Ti合金是互相竞争的过程,将样品暴露于O2氛围中之后,Ti会从合金中分离出来,形成更厚的TiO2覆盖层。这种厚覆盖层在O2中很稳定,表明MSⅠ也可应用于催化氧化反应。
2.3 金属-载体之间的电荷转移过程表征
类似于静态电荷转移的表征思路,基于原位XAFS、气氛条件下的XPS(AP-XPS)等技术,可以考察反应条件下由MSⅠ引起的催化剂表面电性变化[47,53-60]。LⅠU等[47]探究了一种CeO2负载的Ru纳米团簇用于DRM反应的催化活性和稳定性,发现MSⅠ改变了Ru纳米团簇的电子性质,电子转移形成的Ru*-CeO2-x保证了催化活性。在CH4还原气氛中,由原位Ru K吸收边XANES谱图(图9(a))可知,随着温度的提高,RuO2逐渐被还原为Ru,并在150~200 ℃发生了快速还原过程(图9(b))。类似的还原过程也出现在CeO2载体表面,由原位AP-XPS谱图(图9(c))可知,从150 ℃开始,载体表面逐渐产生Ce3+,同时还原过程伴随着氧化物载体中晶格氧的减少。
图9 Ru(NC)/CeO2与CH4反应时的原位Ru K吸收边XANES谱图(a)和对应的拟合结果(b)及6.67 Pa CH4作用下Ru(NC)/CeO2的原位AP-XPS谱图(c)[47]Fig.9 In situ Ru K-edge XANES spectra during reaction of Ru(NC)/CeO2 with CH4 and corresponding fitting results (b) and in situ AP-XPS spectra of Ru(NC)/CeO2 under 6.67 Pa of CH4[47]
同样,通过原位EPR亦能够监测催化反应过程中金属离子的价态变化,进而得到电荷转移的过程信息。如WANG等[55]基于原位EPR辅以一系列非原位表征手段,研究了富氢中CO优先氧化(PROX)反应过程中的CuO-CeO2催化剂的氧化还原行为,认为协同氧化还原是主要的反应机制,即Ce4+被还原为Ce3+伴随着Cu0和Cu+被氧化为Cu2+,这一机制表明了Cu与Ce之间的相互作用对催化性能有重要影响。
需要注意,上述原位表征方法更侧重的是不同反应条件下或实际反应过程中金属-载体之间的电荷转移过程,实际上探测的是电荷转移之后的结果状态(如金属价态)。而真正意义上的电荷转移过程是非常迅速的,其时间尺度一般在飞秒、皮秒量级,这种时间尺度的表征则需要借助时间分辨光谱技术。
本节主要介绍了催化剂预处理条件和反应条件下,MSⅠ可能产生的动态变化,以及表征这类动态性质的常用原位技术,各原位表征方法的特征总结见表2。通过原位表征技术,可以获取特定条件下MSⅠ的强弱和界面微观结构的变化、电荷转移的动态过程等信息,进而理解相互作用和催化反应的机理。目前原位表征的技术和经济门槛均比较高,且大部分原位技术仍然只能满足特定条件下的探测,较难实现任意或实际反应条件下的实时观测,但是随着技术的飞速迭代和发展,原位表征方法将不断成熟和普及。

表2 MSI的原位表征方法总结Table 2 Summary of in situ characterization methods for MSI
3 结语与展望
MSⅠ的宏观概念并不复杂,但是微观细节比较丰富,随着研究者对体系认识的逐渐深入,目前已发展出丰富多样的配套表征方法和策略。事实上,任何一种表征手段都有其特定的使用场景和优缺点,本文根据MSⅠ的不同表现特征对表征技术进行了简单分组,同一种技术经常可以给出不同方面的信息,且MSⅠ的各个特征之间本身也存在内在联系。因此,多种表征手段结合的方式才能实现对体系较全面的认识。
目前,非原位表征方法已经相当成熟,相比原位表征,其使用成本和技术门槛均较低,笔者认为,关于MSⅠ的非原位表征,未来可发展的部分在于更高的空间分辨率,即对微小区域内的组分间相互作用进行表征,如金属原子和化学键的可视化。但是,非原位表征技术只能给出MSⅠ的静态信息,例如预处理或催化反应前后催化剂的形貌、活性金属与载体的价态及电荷分布情况等,而无法表征实际反应过程中相互作用的动态变化。理论上,原位表征技术可以弥补这一缺陷,给出特定条件下MSⅠ强弱的实时变化、电荷转移的动态过程等信息,进而有助于探究MSⅠ的机理和催化反应机理。原位动态表征技术的发展及其在该领域中的应用,还有很长的路要走。因受限于仪器使用条件,如特定压力和温度范围、特定反应气氛和特定反应状态等,目前大部分原位表征技术只能满足特定催化反应的动态表征,因此,如何在实际反应条件下进行原位表征是未来技术需要解决的问题。同时,提高分析方法的时间分辨率,实现真正意义上的动态表征,也是未来研究者们努力的方向。
猜你喜欢
无机化学学报(2023年2期)2023-02-27
华人时刊(2022年9期)2022-09-06
华人时刊(2020年15期)2020-12-14
天然产物研究与开发(2018年5期)2018-06-13
黑龙江工程学院学报(2016年5期)2016-11-12
物理化学学报(2015年7期)2015-12-30
广州大学学报(自然科学版)(2015年4期)2015-12-23
火炸药学报(2014年1期)2014-03-20
影像科学与光化学(2014年3期)2014-03-11
河南科技(2014年12期)2014-02-27
