
儿童2型神经纤维瘤并肝豆状核变性一例报告并文献复习
2024-03-08 06:19许锦平陈先睿姚拥华白海涛
罕少疾病杂志 2024年2期
许锦平 陈先睿 姚拥华 白海涛
厦门大学附属第一医院儿科,厦门大学附属第一医院儿科重点实验室,厦门大学医学院儿童医学研究所 (福建 厦门 361003)
神经纤维瘤病2型(Neurofibromatosis type 2,NF2;OMIM 607379)是因NF2抑癌基因突变所致的一种常染色体显性遗传性疾病,其出生发生率为 1/25000。临床表现主要以双侧前庭神经鞘瘤(Vestibular Schwannomas, VSs)为特征,常伴多发神经系统肿瘤、皮肤及眼等相关病变。病变常累及神经系统有脑(脊)膜瘤、脊髓室管膜瘤、双侧前庭神经鞘瘤(也称听神经瘤)、脊柱肿瘤、其他颅神经鞘瘤和周围神经病变;病变累及到皮肤系统有皮肤或皮下肿瘤和皮肤斑块。常见的眼部病变包括:视网膜错构瘤、白内障及视网膜前膜[1-2]。
肝豆状核变性(Hepatolenticular degeneration,HLD;OMIM 277900)也称Wilson病(Wilson’s disease,WD),是由致病基因ATP7B编码的一种铜转运P型ATP酶功能缺陷或丧失,从而导致胆道排铜障碍,造成大量的铜蓄积于肝、肾、脑、角膜、骨关节等组织脏器的常染色体隐性遗传疾病。因此,临床表现为肝脏损害、肾脏损害、神经精神表现、角膜色素环(Kayser-Fleischer ring,K-F环)及骨关节病等[3-4]。现收治分析1例2型神经纤维瘤并肝豆状核变性的临床特点,并结合文献复习进行分析,以提高对两种疾病的认识及诊疗。
1 临床资料
患儿,女,5岁11个月,因不自主左眼睑下垂4天就诊。4天前无明显诱因出现平视时上眼睑下垂,可遮挡角膜1/3,以3-9点为主,晨轻暮重,无四肢乏力,无口角歪斜,无明显吞咽呛咳,无呼吸困难,无发音异常等不适,2天前眼睑下垂渐加重,渐遮挡角膜2/3,就诊当地医院完善新斯的明试验阴性,考虑“重症肌无力待排”转诊我科神经专科门诊后收入院。本次入院查体:无特殊外貌,精神一般,左侧眼睑下垂,挡角膜3-9点,右眼睑无下垂,活动可,双侧瞳孔等大等圆,对光反应灵敏,心肺腹无异常,病理征阴性。既往史:生后未发现眼睑下垂表现,3年前无明显诱因发现眼球震颤,左侧内斜先后于外省眼科医院行眼震颤手术,1年前当地眼科医院行左眼内斜手术,术后无不适。2个月前眼科医院检查视力右侧0.2,左侧0.75,1月前左侧视力下降为0.05。个人史:G1P1,足月顺产,出生体重3.05kg,无窒息抢救史。出生后予混合喂养。家族史:无类似情况,有一弟弟,3岁,体健。父有1级高血压,母体健,否认近亲结婚。辅助检查:2021.11.08外院眼科:新斯的明试验0.015mg/kg,阴性;胸部CT:胸腺体积增大。头颅CT平扫:未见明显异常。
入院后我院眼科会诊后考虑“诊断:左眼黄斑发育异常,左眼上睑下垂,右眼弱视;建议:戴镜治疗,眼科随访,贵科进一步排除重症肌无力。”,复测新斯的明实验(0.02mg/kg)阴性,生化指标提示“丙氨酸氨基转移酶 200.0 U/L, 天门冬氨酸氨基转移酶 89.0 U/L, 肌酸激酶 113 U/L,乳酸 2.45 mmol/L;血尿串联质谱正常;风湿免疫指标均为阴性;淋巴细胞表型分析正常;腹部B超肝脾未见明显异常声像;脑干视听诱发电位正常;乙酰胆碱受体(AChR,acetylcholine Receptors) 抗体、肌肉特异性受体酪氨酸激酶(MUSK,muscle specific kinase)抗体、低密度脂蛋白受体相关蛋白4(LRP4,lipoprotein-related protein 4)和兰尼碱受体(RyR,ryanodine receport2)均阴性”,予保肝治疗。入院第3天,建议完善遗传基因检测,患儿母亲忆起3年前外院行类似检查,经反复寻找既往资料后基因报告显示“ATP7B基因:位置Exon16(c.3443T>C,p.Ile1148Thr)该变异为错义突变(翻译产物蛋白质第1148位氨基酸残基由Ile变为Thr)、该变异被反复报道为病理性变异;Exon12(c.2804C>T,p.Thr935Met)该突变为错义突变(翻译的产物蛋白质的第935位氨基酸残基上的Thr变为Met),多个研究报道发现Wilson病患者中检测到该突变,可能是中国人群中的热点变异。NF2基因,位置Exon11(c.1009C>T,p.Gln337*),该突变为无义突变(可能使所编码得蛋白质的第337位氨基酸上的Gln变为终止密码子,而提前终止蛋白质翻译)。该突变预计可能造成所编码的蛋白质出现截短从而丧失正常功能。HGMD数据库目前未见相关研究报道;ESP6500siv2_ALL、千人基因组(1000g2015aug_ALL)和dbSNP147数据库也都未见收录。”(见图1),患儿母亲诉未再针对该基因结果进一步诊治随访。遂完善“铜蓝蛋白:79mg/L”以及颅脑MR平扫+增强“1、右眶内视神经外下方、右侧框尖-海绵窦及左侧小脑半球中线旁异常信号灶,左侧Meckel氏腔扩大伴多发强化结节,颅神经多发强化灶(右侧外展神经及双侧三叉神经、听神经、滑车神经、舌咽神经见多个斑点状、线样强化影):考虑神经纤维瘤可能,请结合临床及基因筛查。2、腺样体生理性肥大,左侧下鼻甲肥大。3、颈部多发肿大淋巴结。4、扫描野内附见C4-5水平颈椎管内偏右侧异常强化结节,建议颈椎MR增强。”。诊断明确后,予低铜饮食、口服葡萄糖酸锌片(2.5片,1天3次;元素锌 75mg每天)治疗,同时神经外科会诊后建议进一步完善全脊髓磁共振,家属自动出院转外院进一步诊治。
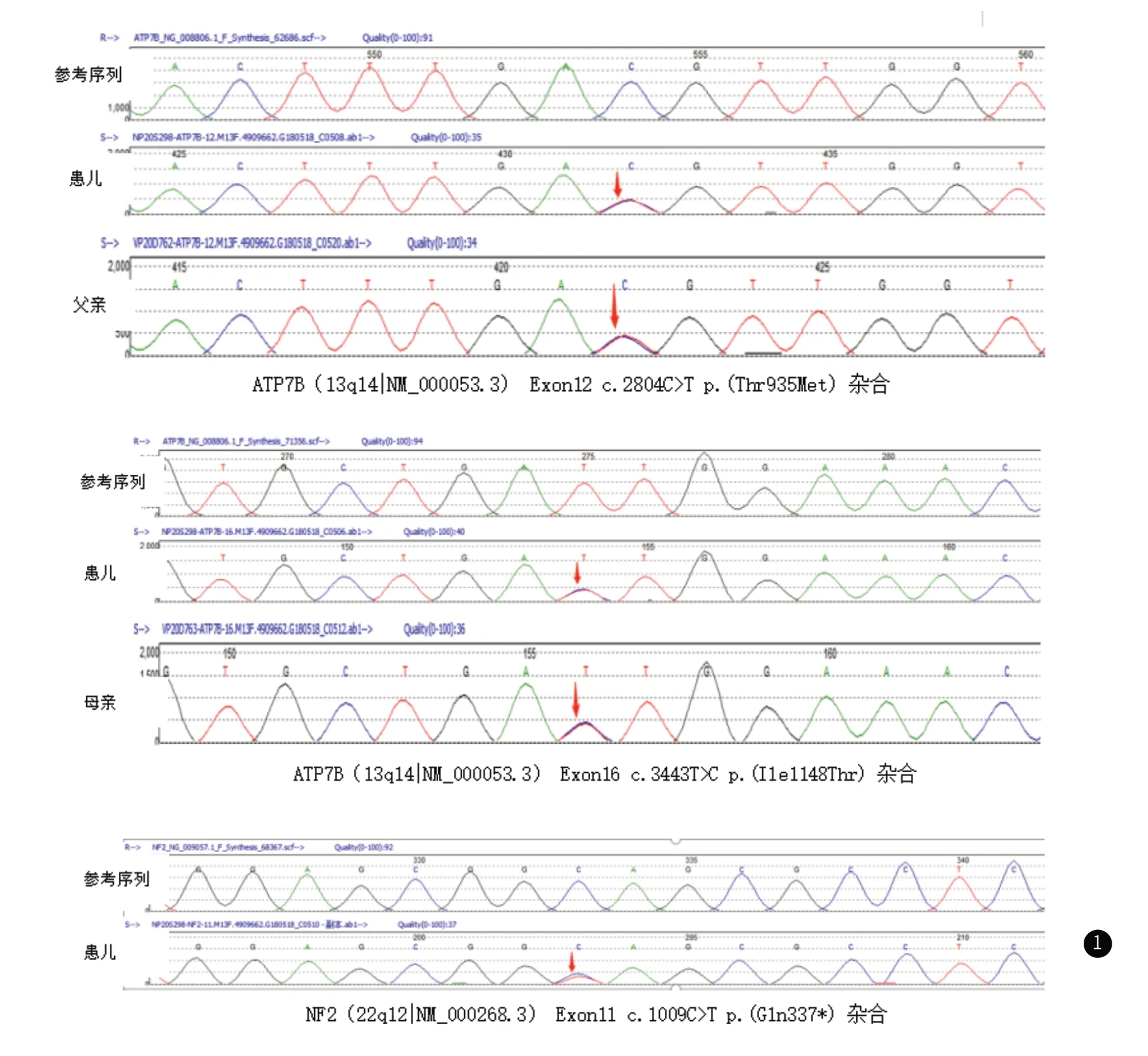
图1 先证者及家系的基因测序图
2 讨 论
神经纤维瘤病 II 型 (NF2) 患病率远低于 1 型神经纤维瘤病,约为 1/60 000。NF2因22q12.2 染色体上的抑癌基因功能丧失引起,该基因长约110000bp,其中编码区cDNA是1785bp,包含的17个外显子中仅前15个外显子具有致病性突变[5]。NF2基因的蛋白产物Merlin(Moesin-Ezrin-Radixin-Likeprotein,又称为Schwannomin)是一种NF2基因编码蛋白,作为肿瘤抑制因子,具有控制细胞生长、增殖和形态有关的细胞信号通路[6-7]。可观察到Merlin在Schwann细胞、脑膜细胞、神经细胞以及晶状体纤维细胞中的高水平表达,其主要通过介导细胞内膜蛋白、肌动蛋白丝以及信号传导效应子之间的相互作用。但Merlin功能缺失以及不同细胞谱系中的细胞内信号传导存在独特而复杂的相互作用,因此,目前尚不完全清楚NF2多样化临床表现的确切机制[8]。
1822年,Wishart[9]最早提出神经纤维瘤病2型,美国国家卫生研究院(NIH)工作组1987年发布共识声明确立了首个神经纤维瘤病 2型诊断标准[10]。2021年,国内共识推荐将“美国神经纤维瘤病2019年会议的修订版”作为NF2的诊断标准,其诊断依据仍以临床表现、影像结果及基因检测为主。该病涉及多个系统,如神经系统中双侧听神经瘤发生率90~95%、脑膜瘤45~58%、髓外肿瘤(脊膜瘤、神经鞘瘤)55~90%、周围神经病变高达66%,眼科疾病中白内障60~81%、视网膜前膜12~40%,皮肤病变中皮肤肿瘤59~68%、皮下肿瘤43~48%,不同NF2患者临床表现可能有较大差异,一般分为重型(Wishart型)和轻型(Gardner型),前者常发病于20岁之前,除前庭神经鞘瘤外常多发颅内及椎管内肿瘤,病情进展较快;后者的发病年龄通常较大(22~27 岁),除听神经瘤外,可不合并其它肿瘤,病情进展较慢[2]。此外,不完全型(Segmental)Wilson病仅表现部分的周围神经系统多发性Schwann 细胞肿瘤,或同侧前庭神经 Schwann 细胞瘤合并脑膜瘤[11]。
肝豆状核变性的世界范围内患病率为1/2 600~1/30 000,携带者频率约为 1/90[1-2]。儿童患者多于5~12岁起病,男女发病比率相似。临床症状早期多样,婴幼儿及儿童多见于肝脏损害,可有3岁起病的肝硬化;神经精神症状多起病于10~30岁,其中肌张力障碍早期多局灶、阶段性,逐渐累及全身;而粗大不规则震颤多为姿势性或动作性;其他系统损害可有肾损害、骨关节病、溶血性贫血等[12]。眼部特征除了角膜K-F环,但7岁内患儿一般无法检出外,其它少见的巩膜黄染、眼睑浮肿、眼球水平震颤、瞳孔对光反射迟钝或消失、复视、眼球运动受限、眼睑下垂等都有相关报道[13-14]。
本例女性患儿,以眼部症状为主要临床表现,2岁时以无明显诱因的眼球震颤,左侧内斜先后于外院行右眼震颤手术及左眼内斜手术,术后动态监测发现视力下降,左侧眼球震颤仍有反复,继而出现上眼睑下垂,反复多次就诊外院中提示ALT和AST轻度升高,未受到重视。此次入院对上睑下垂进行查因分析考虑获得性病因,其中机械性、肌源性、神经肌肉性、神经源性和脑源性上睑下垂[15-16]。患儿早期就有肝酶学轻度升高但无症状,ATP7B基因筛查确定了患儿2条染色体都携带致病突变,符合症状前个体的肝豆状核变性,但未进一步治疗随访;需注意患儿既往眼部症状(眼球水平震颤、眼睑下垂、眼球活动受限)可能与之相关。此次入院乳酸升高,肝酶学异常,注意和线粒体病(如慢性进行性眼外肌瘫痪)进行鉴别。另外,患儿有2次手术病史(上睑提肌腱膜术和结膜-Müller肌切除术),术后眼球震颤无改善,且出现上睑下垂,需注意排查机械性/外伤性引起。患儿外院和我院新斯的明实验及相关抗体检测均阴性,但上睑下垂有晨轻暮重表现,暂不能完全排除,必要时实验性治疗进一步明确。外院颅脑CT未见表现,NF2基因为无义突变且软件预测有害,最终颅脑磁共振显示框内占位病变及多发脑神经瘤样改变(右侧外展神经、双侧三叉神经、听神经、滑车神经、舌咽神经)明确了NF2诊断。回顾国内外文献发现,该患儿为首例 2型神经纤维瘤病合并肝豆状核变性,曾有肝豆状核变性合并1型神经纤维瘤病相关报道[17-19]。
我国肝豆状核变性并不少见,但其可累及多系统致临床表现多样化,学龄前儿童多有肝功能损害和神经精神症状,少见的有肌病、肾损害、白内障以及眼球运动受限、复视、眼睑下垂等眼部非特异性表现[20]。但有研究[21]提示肝豆状的眼球异常运动在垂直方向更多见于水平方向。因此,肝豆状核变性的早期诊断及鉴别仍存在一定困难。由于本病是儿童遗传代谢病中可进行药物有效治疗,且临床预后与治疗时机规范密切相关,2021年国内新指南重点强调了关于症状前个体的早期筛查与干预,并进行终身治疗和终身监测[3]。2型神经纤维瘤病相关肿瘤患者的治疗具有挑战性,由于患者的生存期往往较长,而无法避免疾病所致的临床症状[22]。NF2的相关治疗方式有手术切除、手术减压、药物靶向治疗、立体定向放射治疗和听力康复治疗等。目前更强调的是提高患者生活质量,保存重要神经功能,因此并不一味追求彻底切除肿瘤。本患儿出现上睑下垂,并无肢体瘫痪、平衡障碍等神经功能缺失表现,外院综合评估后暂建议进行密切随访观察。神经纤维瘤病 2型疾病严重程度的主要危险因素有:基因突变位置、突变种类和携带该致病突变的细胞数量。NF2基因型与表型关联的复杂性导致了临床多样化,这决定了每个患者的诊疗方案需要综合评估考虑肿瘤本身的特点,如肿瘤部位、肿瘤大小、肿瘤生长速度、听力损害水平、听力保存情况、肿瘤相关并发症的严重程度以及患者预期生存质量,建议由神经外科、神经内科、人类遗传学、眼科、耳科、听力学、病理科、放疗科、精神科等多学科协作,制定最佳的个体化方案[23-24]。2021年中国专家共识指出NF2非常复杂,无法临床治愈且可能持续进展, 通过多学科协作,在充分评估患者的整体情况、 肿瘤负荷、治疗受益以及治疗风险基础上,合理采用手术、放疗、 药物和神经功能重建等方法制定个体化治疗方案,以期延长患者的生存时间,同时尽可能保护及维持重要的神经功能,达到提高患者的生存质量的目标[2]。
综上所述,本研究中确诊了1例2型神经纤维瘤并肝豆状核变性儿童,发现了一未报道过的NF2基因突变,丰富了人类基因突变数据库。同时,临床医生应对符合症状前个体的肝豆状核变性的患者进行早期识别与治疗随访。此外,关于患儿NF2基因致病性变异的解读以及与临床表型的相关性分析,建议可由临床医师、人类遗传学和基因检测研究机构等相关专家组成疑难病例诊断联盟,进行多学科会诊诊断未明的疑难病例以及后续的遗传分析,以期明确诊断和发病机理,来指导临床治疗[25]。
作者利益冲突:所有均声明无利益冲突
猜你喜欢
中国造纸(2022年9期)2022-11-25
现代临床医学(2021年4期)2021-07-31
阅读(科学探秘)(2021年12期)2021-05-30
环球时报(2019-04-03)2019-04-03
中兽医学杂志(2019年1期)2019-01-06
罕少疾病杂志(2016年4期)2016-03-11
首都医科大学学报(2015年4期)2015-12-16
西藏科技(2015年10期)2015-09-26
中国医疗美容(2015年2期)2015-07-19
中国医疗美容(2015年2期)2015-07-19
