
银-二芳基乙烯化合物的合成及其光致变色性质的研究
2024-03-07 01:33:28李邵蕊徐海兵曾明华
湖北大学学报(自然科学版) 2024年2期
李邵蕊,徐海兵,曾明华,2
(1.湖北省先进有机化学材料协同创新中心(湖北大学),有机功能分子合成与应用教育部重点实验室(湖北大学),湖北 武汉 430062; 2.广西师范大学化学与药物科学系, 广西 桂林 541006)
0 引言
二芳基乙烯开关由中心的乙烯桥和两个芳香基团组成,以优良的热稳定性、抗疲劳性和快速光响应机制成为光致分子开关化合物中一大研究热点。其在一定波长(λ1)的光照下,发生光化学反应,生成另一种构型的产物,再经另一波长(λ2)的照射或热弛豫过程,又可以转换回初始状态[1-3]。通过引入金属模块来调节二芳基乙烯开关的光致变色性能是金属分子开关的一种常见设计理念[4]。金属离子AgI较大的半径和多变的配位构型,使其成为理想的配位中心。通常,银配合物中存在H键、Ag-Ag、Ag-C以及Ag…π等弱相互作用[5],这些相互作用一定程度上能调节配体的结构及其光谱性质。在此,本工作以咪唑为乙烯桥的二芳基乙烯开关L[6]和硝酸银进行配位,得到[L-AgNO3]2(CHCl3)2化合物,简称Ag-L。通过晶体数据分析Ag-L的结构,使用紫外-可见分光光谱仪研究并对比了L和Ag-L的光致变色性质及Ag-L的抗疲劳性。
1 实验部分
1.1 试剂
实验所用原料和氘代试剂均在专业平台购买。除了用于光谱测量的溶剂为光谱级溶剂,所有溶剂在使用前均经过干燥、蒸馏和脱气处理。二芳基乙烯开关L按文献[7]中的方法进行制备。
1.2 表征仪器
使用Bruker傅里叶-红外分光光度计测试红外吸收光谱。核磁氢谱通过400 MHz NMR光谱仪(Bruker Avance 400)进行测试。紫外-可见吸收光谱在Agilent Cary 6000i紫外-可见分光光度计上测定。单晶衍射数据在Rigaku XtaLAB Synergy四圆衍射仪上采集,采用Cu-Kα辐射,使用Cryalispro软件(版本为1.171.39.34b)进行数据还原和分析。利用各向异性位移参数对所有非氢原子进行了细化。结构细化过程中使用的特殊细化如下:由于Ag-L晶体结构中存在无序,i) 使用DFIX指令限制了一些键长;ii) 使用ISOR指令将一些孤立原子限制为近似各向同性。配体L的CCDC号为2257692,配合物Ag-L的CCDC号为2257696,可以通过剑桥晶体学数据中心(http://www.ccdc.cam.ac.uk/data_request/cif)免费获取。
1.3 Ag-L的制备
将二芳基乙烯开关L溶解在乙腈中,加入等摩尔的硝酸银,回流24 h后停止反应,此时溶液为无色澄清溶液,减压浓缩后出现少量白色固体(白色固体为未参与配位的L和过饱和析出的配合物),过滤,取滤液,旋干后得到配合物Ag-L。
2 结果与讨论
2.1 单晶结构描述
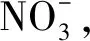

图1 L和Ag-L的晶体结构
比较L配位前后的晶体结构,C5—C6之间的距离在与AgI配位后由原来的0.148 7 nm缩短至0.144 8 nm。配位后吡啶环中C5—N1和咪唑环中C6—N3的键长都发生了变化,这使得C1—N1—C5之间的角度从116.555°变为118.948°,C6—N3—C33之间的角度从106.170°减小为105.984°。L和Ag-L晶体结构的键长与角度变化对比如表1所示。图2展示了不同角度的Ag-L晶体堆积图。此外,我们还发现吡啶环上的质子与相邻苯环之间存在C—H…π相互作用(图2 (a))。

表1 L和Ag-L晶体结构的键长与角度对比
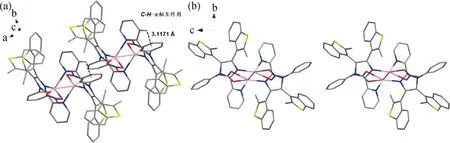
图2 不同角度的Ag-L晶体堆积图
2.2 红外及核磁表征


图3 L与Ag-L的红外光谱对比图
将配体L和配合物Ag-L分别溶于DMSO-d6溶液并置于核磁管中,在室温条件下进行一维核磁共振氢谱的测试。如图4所示,配体L与AgI发生配位后,由于σ效应,L的电子云密度降低,其质子的化学位移明显向低场移动。然而,AgI与N之间存在反馈π键可能会使配体的π系统电荷密度增加,导致配位后部分质子受到了屏蔽作用,向高场移动[10]。

图4 L与Ag-L的核磁氢谱对比图(DMSO-d6, 400 MHz, 293 K)
2.3 光致变色性质研究
为了更好地研究配合物Ag-L的光致变色性质,我们将L和Ag-L分别溶解在脱水处理后的二氯甲烷中,制成浓度为1.0×10-5mol/L的溶液,通过UV-Vis光谱研究它们在累计时间下经365 nm和550 nm光照后发生的光致变色反应。
开环状态下配体L和配合物Ag-L二氯甲烷溶液的UV-Vis谱图中,在300 nm左右时有较强的吸收,溶液呈无色。用365 nm 的紫外光照射后,随着光照时间的增长,L和Ag-L逐渐发生闭环反应,在约550 nm处出现一个新的吸收峰(图5 (a)、(c)),这是闭环异构体的特征吸收,此时溶液颜色由无色变为淡紫色。可见区吸收峰强度随光照时间增长逐渐增强,当光照到一定时间后,吸光度不再变化,此时达到光化学稳定状态(photostationary state, PSS)。紧接着用波长为550 nm的可见光照射,闭环异构体的特征吸收逐渐减弱,直到达到PSS态,溶液由淡紫色变回无色(图5 (b)、(d))。值得注意的是,相比于配位前的L,虽然Ag-L配合物在可见光区的最大吸收峰的波长位移变化较小,但吸收强度有非常大的提升,达到PSS态的时间也从100 s缩短至20 s。综上,引入AgI后,二芳基乙烯化合物在二氯甲烷溶液中仍具有可逆的光致变色性质,但对其光物理和光致变色性能影响较小。

图5 L(a, b)与Ag-L(c, d)在二氯甲烷中吸收随光照时间变化的UV-Vis光谱
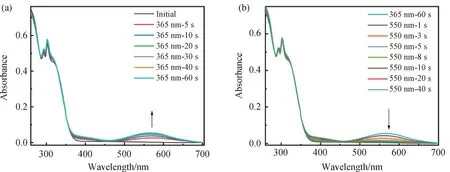
图6 Ag-L(a, b)在PMMA膜中吸收随光照时间变化的UV-Vis光谱
二芳基乙烯化合物的量子产率是评价其性能的一个重要参数。根据监测得到L和Ag-L开闭环过程的吸收谱图,找出开环光稳态与闭环光稳态的最大吸收差值处波长,将该处的吸光度数值作为纵坐标,与之对应的光照时间为横坐标,制作散点图。再根据Y=Y0+Aexp(-kt)方程拟合曲线,得到光致异构过程的速率常数K。再根据公式(1~4),计算光致异构过程的量子产率Φ。其中,Kex是吸收在激发波长处的速率常数,σex为激发波长处的吸收截面,Ψ指的是光通量,I则是激发光源的强度[11-12]。配体L和配合物Ag-L的开闭环量子产率及其相关参数对比如表2所示。

表2 L和Ag-L的开闭环量子产率参数对比
κex=σex×Ψex
(1)
σex= (103× ln(10/NA) ×εirr
(2)
Ψex= 5 × 1015×λirr×I
(3)
Φ=κ/κex
(4)
对配合物Ag-L在聚合物薄膜中的光致变色性质进行探究。首先称取约30 mg的PMMA于30 mL二氯甲烷溶液中,加热使PMMA溶解,将约1 mg的Ag-L加入到完全溶解的PMMA溶液中,搅拌均匀。最后从中取适宜体积的溶液于干净平整的玻璃板上,使溶液冷却、溶剂挥发,制成薄膜,进行UV-Vis光谱测试。
处于PMMA薄膜中的配合物Ag-L,初始状态呈白色,在波长为350 nm之前有很强的吸收,经365 nm的紫外光照射后,发生闭环反应,在570 nm 的位置出现新的吸收峰。该吸收峰随光照时间的增长吸收强度不断增大,照射60 min时达到PSS态,薄膜颜色从白色变为紫色。接着用波长为550 nm 的可见光照射,570 nm处的新吸收峰强度逐渐减弱,直到照射40 s后回到初始状态,此时薄膜由紫色变回无色。相对于在二氯甲烷溶液中,i) PMMA中的Ag-L经紫外光照闭环后,闭环状态的特征吸收出现轻微红移,吸收扩展到了700 nm左右,这表明聚合物基质对配合物Ag-L的光致变色性质造成了影响。ii) Ag-L在PMMA膜中由于受到高分子介质的高粘度影响,限制了其在开闭环两个状态之间的自由转换[13]。其中,开环反应在此介质中受到的影响较大,开环反应的时间从5 s延长至40 s。
2.4 抗疲劳性研究
通过UV-Vis光谱分别对配合物Ag-L在二氯甲烷溶液和PMMA膜中的光致变色过程的抗疲劳性进行研究。以初始开环状态和365 nm辐照闭环为第一个循环,随后通过550 nm辐照开环后再用365 nm照射为第二个循环,不断重复光化学闭环和开环反应[14],绘制最大吸收波长处的吸光度随循环次数变化关系的曲线。如图7 (a)所示,配合物Ag-L的二氯甲烷溶液的光致变色性能随循环次数的增加而缓慢衰减。相比于L[6],Ag-L在二氯甲烷中由于经受反复长时间辐照的使得金属和配体之间的配位作用减弱,出现了少量配合物分解的状况。而Ag-L的PMMA膜经过约10次开闭环状态的转化后(见图7 (b)),闭环状态下最大波长处的吸收没有发生较大变化,这表明配合物Ag-L的抗疲劳性较好,具备一定的应用于光学信息存储能力。

图7 Ag-L在DCM溶液(a)和PMMA膜(b)中光致异构的抗疲劳性研究
3 结论
本工作合成了一种银-二芳基乙烯化合物[L-AgNO3]2(CHCl3)2。运用单晶衍射仪、红外光谱仪及核磁共振波谱仪对Ag-L结构进行了分析。在该化合物中,AgI同时与两个不同分子上的两个N原子结合,形成了一个扭曲的四面体。通过UV-Vis分光光谱仪对Ag-L的光致异构过程进行研究,研究表明配位后其仍然具有良好的可逆光致变色性能,并且可以通过在不同介质中调节其开关环反应速率。这为开发光子型储存材料提供了可参考设计理念和研究思路。
猜你喜欢
上海化工(2018年10期)2018-10-31 01:21:06
电子测试(2018年1期)2018-04-18 11:52:24
合成化学(2015年2期)2016-01-17 09:04:21
合成化学(2015年4期)2016-01-17 09:01:04
化工进展(2015年6期)2015-11-13 00:27:23
海军航空大学学报(2015年1期)2015-11-11 17:22:41
中国塑料(2015年10期)2015-10-14 01:13:13
化工生产与技术(2014年5期)2014-02-27 13:42:02
东南大学学报(自然科学版)(2012年3期)2012-06-28 03:59:00
河北大学学报(自然科学版)(2012年3期)2012-03-25 10:13:01
