
Genome-wide association with transcriptomics reveals a shade-tolerance gene network in soybean
2024-03-07 01:50YnzhuSuXioshuiHoWeiyingZengZhengungLiYongpengPnCnWngPengfeiGuoZhipengZhngJinoHeGungnnXingWuinWngJiopingZhngZudongSunJunyiGi
The Crop Journal 2024年1期
Ynzhu Su, Xioshui Ho, Weiying Zeng, Zhengung Li, Yongpeng Pn, Cn Wng,Pengfei Guo, Zhipeng Zhng, Jino He, Gungnn Xing, Wuin Wng, Jioping Zhng,Zudong Sun,*, Junyi Gi,*
a Soybean Research Institute/MARA National Center for Soybean Improvement MARA Key Laboratory of Biology and Genetic Improvement of Soybean/State Key Laboratory for Crop Genetics and Germplasm Enhancement/State Innovation Platform for Integrated Production and Education in Soybean Bio-breeding/Jiangsu Collaborative Innovation Center for Modern Crop Production, Nanjing Agricultural University, Nanjing 210095, Jiangsu, China
b Guangxi Academy of Agricultural Sciences, Nanning 530007, Guangxi, China
Keywords:Soybean (Glycine max (L.) Merr.)Shade-tolerance Restricted two-stage multi-locus genomewide association study based on gene/allele sequence markers (GASM-RTM-GWAS)Shade-tolerance index (STI)Relative cell length (RCL)Transcriptome
ABSTRACT Shade tolerance is essential for soybeans in inter/relay cropping systems.A genome-wide association study(GWAS)integrated with transcriptome sequencing was performed to identify genes and construct a genetic network governing the trait in a set of recombinant inbred lines derived from two soybean parents with contrasting shade tolerance.An improved GWAS procedure, restricted two-stage multi-locus genome-wide association study based on gene/allele sequence markers (GASM-RTM-GWAS), identified 140 genes and their alleles associated with shade-tolerance index(STI),146 with relative pith cell length(RCL),and nine with both.Annotation of these genes by biological categories allowed the construction of a protein–protein interaction network by 187 genes, of which half were differentially expressed under shading and non-shading conditions as well as at different growth stages.From the identified genes,three ones jointly identified for both traits by both GWAS and transcriptome and two genes with maximum links were chosen as beginners for entrance into the network.Altogether, both STI and RCL gene systems worked for shade-tolerance with genes interacted each other, this confirmed that shadetolerance is regulated by more than single group of interacted genes, involving multiple biological functions as a gene network.
1.Introduction
An innovative maize–soybean inter/relay cropping system has been recognized as an approach to alleviating the shortage of soybean acreage in China [1].This system, designated maize–soybean composite strip inter/relay cropping, is characterized by alternating wide and narrow maize row spacing to obtain maize yield similar to that of monocropping plus an additional harvest of soybean[2,3].
Shading by maize in this cropping system impairs soybean morphological and anatomical development.It increases hypocotyl length [4],blade inclination [5], petiole, internode, and plant length[6],and stalk lodging and vining[7]and reduces specific leaf weight and leaf area per plant [5,8], stem diameter[6], root/shoot ratio [7],number of pods and seeds, and seed yield [9].As a shade-tolerance index (STI), Sun et al.[10] proposed the mean of the ratios of plant height and internode length on the 50th day after sowing under 30%shading to their respective values under non-shading,the smaller the STI, the greater was the shade tolerance.In addition to the external morphology indicator, the internal anatomy indicator should be considered, such as cell length of epidermis, phloem,xylem, and pith, which is correlated with stem length elongation and lodging score [11].Cell-level morphological and anatomical traits can be measured by micrography and micrometry [12].To choose one internal indicator, the relative pith cell length (RCL) on main stem under shading versus non-shading stress was suggested.Thus, STI integrated with RCL will be used to explore jointly the genetic system of shade tolerance in this study.
Quantitative-trait loci (QTL) and genes controlling quantitative traits may be associated with DNA markers by linkage or genome-wide association mapping (GWAS).Linkage mapping by approaches such as composite interval mapping (CIM) [13] is employed in biparental populations such as recombinant inbred line (RIL) sets derived from a pair of diverse parents, and GWAS is used for panels of unrelated germplasm accessions.
A GWAS variant, restricted two-stage multi-locus model GWAS(RTM-GWAS, https://github.com/njau-sri/rtm-gwas or https://gitee.com/njau-sri/rtm-gwas) with single-nucleotide polymorphism(SNP) linkage disequilibrium blocks (SNPLDB) used as genomic markers [14–18], has been used to identify QTL in RIL sets, where population structure bias is lower than that in germplasm panels.In a soybean RIL population, QTL for days to flowering detected by RTM–GWAS detected almost all loci identified by CIM and another GWAS variant, mixed linear model GWAS, and accounted for the genetic proportion of phenotypic variation predicted by the heritability of the trait [16].RTM–GWAS achieved similar success with seedling drought tolerance [17] and seed-flooding tolerance [18].In our previous study [19], a gene/allele sequence marker (GASM,an allele sequence segment of a gene, or simply a set of allele SNP(s) of a gene) identified by whole-genome sequencing was used to replace SNPLDB in RTM-GWAS (named GASM-RTM-GWAS) for shade tolerance in a southern Chinese soybean germplasm panel.It identified 63 candidate genes with 318 alleles and their effects.
In view of the lack of genetic studies of soybean shade-tolerance in inter/relay cropping systems,we proposed to identify genes and gene networks governing the trait.The experimental population was a biparental set of RILs, the external morphological indicator was STI,and the internal anatomical indicator was relative pith cell length (RCL) under shade stress vs.non-stress.Candidate genes were to be identified by both GASM-RTM–GWAS and gene expression variation in response to shading.The objectives were to (i)Characterize the variation of morphological STI and anatomical RCL under shade-stress vs.non-stress in the RIL population, (ii)explore the STI and RCL gene and their allele systems, and predict recombination potential for breeding purpose of the population,(iii) construct a model gene network describing shade-tolerance gene functions and their interactions, (iv) explore the features and relationships between GWAS vs.transcriptome analysis of shade-tolerance systems, and (v) select key genes as entry points for further studies on shade-tolerance gene network.
2.Materials and methods
2.1.Plant materials and field experiments
Two soybean parents selected from the SCSGP, tolerant Aaijiaozao and sensitive Chippewa,were crossed and an RIL population(AC-RIL) of 246 F2:5:7lines (F2:5-derived lines in F7) was developed.The RILs and parents were grown at the experimental station of the Guangxi Academy of Agricultural Sciences, Nanning, Guangxi,sown on April 1, 2017, April 6, 2018, and March 28, 2019.In each season,two sets of experiments were arranged in comparable conditions: one set shaded with plastic film to cause 30% light reduction and another in an open field as a control[9].Both sets were arranged in the same randomized complete block design with treatment and check plots of each accession situated in similar locations, so that calculated STI values would be as comparable as possible.Each set was replicated three times, with one-row plots, 1.2 m in length,0.45 m apart, and 13 plants per row.Plant height (PH) and main stem node number were recorded for five consecutive middle plants on the 50th day after sowing.The STI was calculated as follows:
where PHshadingand AINLshadingare plot means,PHckand AINLckare means of three replicates, and AINL is mean internode length[10,19].
2.2.Anatomical observation of penultimate internode on the main stem
On the 50th day after sowing in 2018, the middle segment of the penultimate internode on the main stem was sampled from three plants in each treatment and control plot.The segments were immersed in a formaldehyde-acetic acid-ethanol fixative (38%formaldehyde/glacial acetic acid/70% alcohol, 5:5:90, v/v/v) for 20 min, dehydrated in a graded (70%–100%) ethanol series, vitrified with TO-type biological tablet transparent agent (Cenxi, Guangxi,China) graded with ethanol (1:2 TO:ethanol v:v to 100% TO),embedded in paraffin, and cut into 12-μm sections with a rotary microtome(RM2255,Leica Microsystems Ltd.,Wetzlar,Germany).They were stained with safranin O and counterstained with fast green.Pith cell length was recorded under a light microscope(Nikon Ni-E, Yokohama, Japan).
To measure pith cell length, a longitudinal section specimen was randomly selected,and 8–10 micrographs were captured,with all pith cell lengths measured in each micrograph using the software NIS-Elements D 4.11.00 (Nikon Corporation, Japan).RCL values were calculated for each plot of the RILs and parents, as
where CLshadingis the mean length of all pith cells in the same micrograph under 30% shading and ACLCKis the mean length of all pith cells (over multiple photos) of the control.Owing to the labor intensiveness of the anatomical measurement,five RCL values for each RIL in only one replicate were randomly selected for statistical and association analyses.
2.3.Statistical analysis
Descriptive statistics and ANOVA of STI and RCL were calculated using PROCs MEAN and GLM in SAS software (version 9.4; SAS Institute Inc.,Cary,NC,USA).ANOVA was performed by constructing a linear model with line, environment, replication, and genotype × environment as random effects on STI, and line as a random effect on RCL (one-way random design).The population heritability (h2) was calculated from the expected least squares as h2= σ2g/(σ2g+ σ2ge/n + σ2ε/nr) for STI and h2= σ2g/(σ2g+ σ2ε/r) for RCL, where σ2g,σ2ge, and σ2εare genotype, genotype-by-environment interaction,and random error variance,respectively;n is the number of environments,and r is the number of replications[20].Genotypic coefficient of variation(GCV)was calculated as GCV=σg/μ,where μ is the estimated population mean [21].
2.4.Genotyping of the AC-RIL population
Whole-genome resequencing based on Wm82.a2 in SoyBase(http://www.soybase.org) for the AC-RIL population and parents was performed with an Illumina HiSeq 2000 instrument(Annaroad Gene Technology(Beijing) Co., Ltd., China) on DNA extracted from fresh young leaves by the cetyltrimethylammonium bromide method [22].The sequencing depth was ~2.0× for the RILs and~5.0× for the parents.All sequence reads were aligned against the reference genome Wm82.a2 in SoyBase using BWA v0.7.9a[23] with default parameters.SNP calling was performed with Genome Analysis Toolkit (GATK; version 3.3.0) [24]and SAMtools[25].To obtain high-quality SNPs, first, SNPs with the same genotype in both parents were filtered out, and filtered SNPs were tested by chi-square (χ2) to confirm that the genotypes met the 1:1 segregation ratio.Then, genotypes were filtered for a missing rate ≤0.3.Finally, npute-lnx64 software [26] was used to fill in the missing SNPs.After another χ2test, 33,170 SNPs were identified in the AC-RIL population.
SNPs in a gene region were assembled into a GASM using RTMGWAS.The start and end positions for each GASM were those of one of the 53,625 genes in the reference genome Wm82.a2.A few nonparental haplotypes or alleles were replaced with the most similar parental haplotype or allele as previously described [14].
2.5.Identification of STI and RCL gene systems in AC-RIL using GASMRTM-GWAS and SNPLDB-RTM-GWAS
Genes associated with STI and RCL,and their alleles,were identified using GASM-RTM-GWAS.At the first stage, simple linear regression based on single-locus model with significance level at P ≤0.05 was employed to preselect GASMs.Then, at the second stage, stepwise regression characterized with forward selection and backward elimination under a multi-locus model with and without gene × environment interaction (GEI) for STI and RCL,respectively, was applied to the preselected GASMs to identify associated genes at multi-locus model P ≤0.05.That is a similar model like the Bonferroni correction P/m (P is significance under single-locus model 0.05, m is the marker number).A gene with 1.50% or greater contribution to PV was designated as a largecontribution(LC)major gene,and that with less than 1.50%contribution to PV as a small-contribution (SC) major gene.The boundary between the two set of genes is around the inflection point of the distribution curve of gene contribution, which is not fixed,but varies among curves.Genes identified and their corresponding allele effects were arranged into separate gene-allele matrices for STI and RCL, respectively.
The operation method of SNPLDB-RTM-GWAS procedure is the similar as GASM-RTM-GWAS, except SNPLDB is defined as SNP linkage equilibrium block, which can be referred to He et al.[14].
2.6.Gene function annotation and shade-tolerance protein–protein interaction network construction
The STI and RCL genes identified were annotated with their functions using SoyBase based on the reference genome Wm82.a2.To identify gene interaction in the shade-tolerance gene system,protein sequences of the genes identified for STI and RCL were obtained from the Phytozome database(http://phytozome.jgi.doe.-gov) and then submitted to the ‘‘Glycine max” section of the STRING database Version 11.5 (http://string-db.org, last accessed August 23, 2022) [27] to generate protein–protein interaction(PPI) networks.The ‘‘minimum required interaction score” is the approximate probability that a predicted link between two proteins exists.The recommended confidence limits are as follows:low confidence 0.15, medium confidence 0.4, high confidence 0.7,highest confidence 0.9, and custom value (confidences in 0–1), or choosing a value based on a specific objective (http://string-db.org).Because we wished to identify interacting-gene groups large enough that the identified hubs would be broadly representative,we accepted a confidence score of 0.15.In the PPI network, nodes(genes) with ≥20 links were assigned as key hub genes.
2.7.Transcriptome analysis
From plants of the parents Aijiaozao (A) and Chippewa (C),grown under shading (T) and non-shading (CK) conditions, 3–5 main-stem tips were sampled on day 30(when growth differences had appeared) and day 40 (when growth differences had increased) for RNA sequencing on an Illumina HiSeq2500 instrument (Gene Denovo Biotechnology Co., Guangzhou, Guangdong,China).These samples were numbered AT30, ACK30, CT30, CCK30,AT40, ACK40,CT40,and CCK40.Raw read filtering,gene mapping,gene expression abundance calculations, and differential gene expression analysis were performed as described previously [28].Genes or transcripts with P ≤0.05 and |fold change|≥1.5 were assigned as differentially expressed genes (DEGs) [29] using DESeq2 software [30].Under shading and non-shading conditions, three biological replicates were sequenced for each parent on day 30, but only one replicate for each parent on day 40.DEGs were annotated and functionally classified using Kyoto Encyclopedia of Genes and Genomes (KEGG) [31].Pathways meeting false discovery rate <0.05 were defined as significantly enriched pathways in DEGs[32].The expression levels of two randomly selected DEGs(Table S1) for each parent were measured using SYBR Green PCR Master Mix solution (TakaRa Bio.Inc, Dalian, Liaoning, China) on a CFX96 Touch Quantitative Real-Time PCR Detection System(Bio-Rad, Hercules, CA, USA) with TUB4 (ribosomal-ubiquitin fusion protein)as internal control.Fold changes in gene expression were calculated as described by Zhao et al.[33].
2.8.Construction of a linkage map and CIM
CIM was employed to find QTL using WinQTLCart v2.5 software(https://brcwebportal.cos.ncsu.edu/qtlcart/WQTLCart.htm) [34].A GASM genetic linkage map containing 20 linkage groups (representing chromosomes) was constructed using JoinMap 4.1 (www.kyazma.nl) with a minimum logarithm of odds (LOD, the commonly used logarithmic value for the probability ratio of linkage and non-linkage between two genetic loci) value of 3 (LOD ≥3,the probability of linkage between two loci is more than a thousand times higher than that of non-linkage).In CIM, 1000 permutation tests at a 95% confidence level were used to set the LOD threshold.A minimum LOD value of 2.5 was used to indicate the presence of a QTL [35].
3.Results
3.1.Characteristics of STI and RCL in a tolerant × sensitive RIL population
Under shading stress, Chippewa and sensitive lines exhibited slenderness, bending, overgrowth, and lodging with increased plant and internode length, whereas Aijiaozao and tolerant lines showed little morphological change and remained upright(Fig.1A).
In Fig.1B,the cells of the cortex,phloem,xylem,and pith of the shade-sensitive parent and lines are elongated in the longitudinal section under shade stress, causing the increased plant height,whereas those of the shade-tolerant parent have changed little under either shading or non-shading conditions.In the longitudinal and cross sections, the tissues of the tolerant Aijiaozao and lines are relatively well developed and closely arranged under shading.However,the cell layers of the cortex,phloem,and xylem of the shade-sensitive Chippewa/lines have decreased and are severely degraded, causing decreased stem diameter, unclear structure, small cells, and difficulty in distinguishing them, but with the pith tissue accounting for a wide area of the whole section with large and clear parenchyma cells (Figs.1B, S1).This explains why the pith cell length was focused on.
STI and RCL varied among the RILs, and STI varied with RILs ×environments (Table S2, single environment for RCL).In AC-RIL population, both traits showed transgressive segregation at both directions (Table S3), the STI of the RILs varied less wider than RCL (1.29–2.50 vs.0.48–3.68) (Table S3).We have found some excellent lines in transgressive progenies, such as the lines RAC165 (STI 1.33, RCL 0.97) and RAC166 (STI 1.32, RCL 0.90).This finding means that the genetic structure of the parents is complementary to both STI and RCL, which provides the genetic basis for shade-tolerance improvement in AC-RIL population.

Fig.1.Schematic diagram of soybean stem under natural light and shade stress.(A) Growth state of two parents and three RILs under sunlight (CK) and 30% shading (T).Leaves have been trimmed off to facilitate observation of plant stem growth.PH is plant height; AINL is average internode length.(B) Longitudinal and cross section of the penultimate internode on the main stem of the two parents and two lines under shading and non-shading 50 d after sowing(10×magnification).ACL is the average length of all pith cells.
The genotypic coefficients of variation and heritability were estimated as respectively 12.95% and 97.48% (with h2g×eincluded;72.71% if h2g×ewas not included) for STI and as high as 31.26% and 98.28% for RCL (in only one environment, so that h2g×ecould not be estimated) (Table S3).STI and RCL were correlated (r = 0.45,P < 0.001, Table S3).
3.2.Identification of STI and RCL gene systems using GASM-RTMGWAS
A total of 2919 GASMs were obtained on the 20 chromosomes each with 37–393 GASMs.STI and RCL genes identified using GASM-RTM-GWAS are described in Tables 1, S4 and S5, and Fig.2.For STI, 140 genes were identified, including 105 maineffect genes and 100 gene × environment interaction (GEI) genes,accounting for 91.96% (60.36% of main-effect genes) of the total phenotypic variation (PV).For RCL, 146 main-effect genes were detected,explaining 95.59%of PV,without GEI genes because only a single environment was used.Nine genes were associated with both STI and RCL (Fig.2B).Among the identified genes, 7 STI and 15 RCL genes were LC ones, contributing a total of 17.78%(1.87%–3.33%) and 34.97% (1.53%–4.16%) PV, respectively, and the remaining 98 and 131 ones were SC genes,contributing a total of 42.58% (0.08%–1.26%) and 60.62% (0.04%–1.49%) PV (Table 1),respectively.Both LC and SC genes in STI and RCL were conferred by numerous genes with continuously varying PV contributions(Fig.2C; Table 1).In the two genetic systems, the total maineffect genetic contributions (h2) were respectively 72.71% and 98.28% for STI and RCL, and the remaining variances, 72.71% -60.36% = 12.35% and 98.28% - 95.59% = 2.69% could be due to unmapped minor genes or polygenes, including some influence from intergenic elements (Table 1).The GEI effect is an important property for the STI gene system, but 65 of the 100 GEI genes also functioned as main-effect genes, and the environmental factor‘‘year” was not fixed (Table S4).
The 105 and 146 main-effect genes for STI and RCL,respectively,contained two parental alleles each, and the absolute value of the allele effects ranged from|0.56|to|1.31|(Fig.2D;Tables S6,S7).All the detected genes with their alleles and effects on each indicator were organized into a gene-allele matrix as a simplified form of the genetic structure of the AC-RIL population, as well as that of each line, with the genetic constitutions of the two parents included(Fig.2E).The matrices show that all the RILs contained both positive and negative alleles.
From the gene-allele matrices,the breeding potential of STI and RCL for pure lines was predicted.Under the linkage and independent assortment models, the 15th percentile values (shadetolerant direction) were 0.58 and 0.13 (linkage) and -0.15 and-3.70(independent)for STI and RCL,respectively,while the corresponding observed values of the RILs were 1.29 and 0.48 and those of the tolerant parent were 1.38 and 1.20, respectively.Additionally, if the elite (negative) alleles at all loci converged the extreme genotypic values will be -13.77 and -46.22 for STI and RCL,respectively (Table S8).The great difference of STI and RCL between the two prediction models means a tight linkage existed in the two genetic systems,there are potential to be released from further recombination.Especially.the high transgression may be used in shade-tolerance breeding programs.
3.3.The STI and RCL gene systems are interrelated as a joint shadetolerance gene network
The 140 STI and 146 RCL genes were grouped into the same six categories (Fig.3A; Table S9, summarized in Tables S6, S7): category I, 12 STI and 5 RCL signal transduction genes; category II, 17 STI and 11 RCL transport genes; category III, 23 STI and 23 RCL metabolic process genes with two shared genes; category IV, 28 STI and 28 RCL gene expression related genes; category V, 31 STI and 51 RCL catalytic activity genes; and category VI, 29 STI and 28 RCL unknown biological pathway genes with one shared gene.Category V had the most STI and RCL genes,followed by categories III, IV, and VI, whereas categories I and II had the fewest genes,showing that many enzymes become very active under shade stress.The PV of categories III and VI was larger for STI (14.51%and 17.78% PV), and that of categories V and VI was larger for RCL (36.62% and 17.29% PV).However, the PV of categories IV and V in RCL was much larger than that in STI,indicating that most STI and RCL genes have similar, even identical functions, but that the genes in the functional pathways regulating the two traits still differed (Tables S6, S7, S9).
Based on the above similarities between the two sets of genes,the PPI networks were predicted to explore the potential relationship between STI and RCL gene systems.Of the 277 STI+RCL genes(with nine shared genes), 195 were predicted to form five PPI groups, that is, eight genes (Fig.3C–F) into four groups with two genes each, whereas the remaining 187 genes were classified into the largest group in a highly complex linked manner, each hub linked to both STI and RCL genes (Fig.3B).There were 99 STI and 101 RCL genes with five shared ones and 40.94%and 66.60%contributions to PV, respectively.The gene network is composed of different functional genes, the 195 (70.4%) of the 277 ones in the PPI networks involved in all the six functional categories:category I (7 STI + 3 RCL), II (15 + 8), III (19 + 21), IV (21 + 21), V (25 + 40),and VI(12+8).This shows that the shade-tolerance gene network is a composite of both STI and RCL genes with their functions interrelated as a uniform entity(Table S9).The key hub genes were distributed in functional gene categories II (2), III (1), IV (4), and V(11); however, none was found in categories I and VI.As shown in Fig.3B, 18 key hubs were identified with the largest ones of gST15.05 (79 links) and gRCL15.13 (95 links), wherein 13 key hubs(encoding helicase,polymerase,and ribosomal proteins,Tables S6,S7) were involved in DNA replication, transcription, and translation, which are cores for life-sustaining activities and respond to abiotic stress.
Although the STI and RCL gene systems are in a joint shadetolerance gene network, they are two different gene systems.Thedifferences between the two systems are as follows:(i)The phenotypic variation of RCL was wider and more sensitive to shade stress.(ii) More RCL genes, especially large-contribution genes(15 of 146, such as gRCL04.05, gRCL10.06, and gRCL15.03),were identified than STI genes (7 of 140, such as gST14.08).(iii)Importantly,one gene (gRCL13.03) involved in integral membrane components,two genes(gRCL06.12 and gRCL11.01)involved in cell division,and nine genes(e.g.,gRCL09.01)involved in synthesis and degradation of cell wall macromolecules in RCL, but not in STI(Tables S6,S7).(iv)The RCL genes(such as gRCL15.13)were located at the most central position of the PPI network, indicating an anatomical trait with more basic and broad links.

Table 1 STI- and RCL-associated genes in the AC-RIL population.
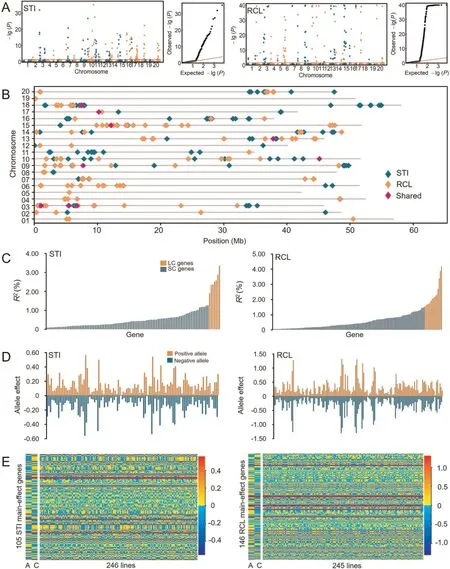
Fig.2.Identification of gene-allele system conferring shade-tolerance in the AC-RIL population.(A)Manhattan and quantile-quantile plots for shade-tolerance index(STI)and relative pith cell length(RCL)using gene-allele sequence markers in RTM-GWAS.-lg P>40 are shown as 40 in RCL.(B)The distribution of genes associated with STI and RCL on 20 soybean chromosomes.(C)The phenotypic contribution of the 105 STI and 146 RCL main-effect genes detected.The horizontal axis indicates the genetic contribution to phenotypic variance(R2,%),arranged in increasing order.(D)The allele effects of STI and RCL genes.The dark yellow and green bars represent positive and negative alleles,respectively.(E) The STI and RIL gene–allele matrix of the parents and RIL populations.The horizontal axis shows 246 or 245 RILs arranged in increasing order from left to right for STI and RCL values.The vertical axis shows the allele effects expressed in colors, arranged in increasing chromosome order from bottom to top, where yellow indicates a positive value, blue indicates a negative value, and color darkness indicates larger absolute value.

Fig.3.The functional category (groups) and the PPI network for STI and RCL genes.(A) The functional category/groups of STI and RCL genes.The numbers I–VI at the top indicate the six functional categories of STI and RCL genes.Category I is signal transduction genes; category II is transport genes; category III is metabolic process genes;category IV is gene expression-related genes; category V is some genes participating in catalytic activity; and category VI is genes with unknown functions.The numbers below I–VI represent the R2 of each group of main-effect genes.The numbers above the histograms are the number of genes corresponding to each group(see also Tables S6 and S7).‘‘CW enzymes”and‘‘NAM enzymes”in the abscissa represent enzymes of respectively carbohydrate metabolism and nucleic acid metabolism.The light blue fonts in the abscissa correspond to the same functional groups in STI and RCL.(B–F) Five protein–protein interaction (PPI) networks of STI and RCL genes in the AC-RIL population.Each node represents a gene(protein).Nodes of the same color belong to the same gene function category.Some large nodes on the outermost circle layer are key hub genes with links ≥20.
3.4.Expressed gene system associated with response to shade stress
The RNA-seq data presented in Table S10 suggest a high accuracy of sequencing,and the quantitative real-time PCR results were consistent with the transcriptome (Fig.S2A).In transcriptomic analysis of the two parents using specimens collected on day 30 under shaded and non-shaded environments, a total of 32,111 expressed genes were revealed (Fig.S2B).The four comparison groups, AT30vs.ACK30, ACK30vs.CCK30, AT30vs.CT30, and CT30vs.CCK30with 2649, 3807, 6577, and 6561 significant DEGs(Fig.S3A), respectively, showed differences in expression levels not only between materials but between treatments.In total,11,771 DEGs (with duplicates removed) were identified in all four pairs of comparisons.The DEGs between shading and non-shading of the two parents (AT30vs.ACK30and CT30vs.CCK30) were 1421(761 + 234 + 322 + 104) and 5333 (2200 + 2147 + 542 + 444),respectively, plus 1228 (307 + 240 + 95 + 586) shared DEGs(Fig.4A).The DEGs between AT30vs.CT30and ACK30vs.CCK30also belong to the DEGs between shaded and non-shaded environments and were 1700(927+234+95+444)and 4470(1708+2200+322+ 240), respectively, plus 2107 (1154 + 104 + 307 + 542) shared DEGs.Because many DEGs were common to the two pairs of comparisons,the total number of DEGs associated with the shade treatments was 9389([1421+5333]+[927+1708]or[1421+5333]+[1700 + 4470] – [234 + 95 + 444 + 322 + 240 + 2200]) (Fig.4A).

Fig.4.DEGs and KEGG pathway enrichment for the two parents.(A) Transcriptomic analysis of the two parents on days 30 (left) and 40 (right), and DEGs coincide with GWAS-identified genes(middle).In the Venn diagrams of day 30 and 40 samples,11,771 and 20,110 from a total of 21,476 DEGs were identified from the differential response between the two parents, in which 22 + 29 = 51 and 43 + 29 = 72 from a total of 94 DEGs were also identified from GWAS.(B) Classification of the DEG functions corresponding to shade and non-shade conditions using KEGG database.At the first level classification, five categories of DEGs are included (total seven categories in KEGG)” ‘‘Metabolism”, ‘‘Genetic Information Processing”, ‘‘Environmental Information Processing”, ‘‘Cellular Processes”, and ‘‘Organismal Systems”; at the second level, 19 groups are included(colored fonts in the figure),such as carbohydrate metabolism;at the third level,the pie chart shows that the carbohydrate metabolism group involves 15 metabolic pathways.Here, the ID of this pathway in KEGG is ko00660.DEG, differentially expressed gene; KEGG, Kyoto Encyclopedia of Genes and Genomes.
To identify biological pathways active under shade conditions,all DEGs responding to shade stress were classified by reference pathways in the KEGG database.A total of 2800(29.8%,with some genes having multiple functions) DEGs were categorized into five KEGG main categories and 19 subcategories(Fig.4B).Among these pathways, ‘‘Metabolism”, including ‘‘Carbohydrate metabolism”,had the most DEGs (567, Fig.4B).Most of the DEGs in the other four categories were markedly enriched in subcategories‘‘Folding,sorting, and degradation” (119), ‘‘Signal transduction” (246),‘‘Transport and catabolism”(119),and‘‘Environmental adaptation”(105).The functional categories (I–VI) of GWAS-identified genes were involved in the following pathways:category I corresponded to ‘‘Signal transduction” in ‘‘Environmental Information Processing”; category II in ‘‘Membrane transport” and ‘‘Transport” in‘‘Environmental information processing” and ‘‘Cellular processes”,respectively; and category III in‘‘Catabolism”and‘‘Multiple metabolism (carbohydrate metabolism, nucleic acid metabolism)” in‘‘Cellular processes” and ‘‘Metabolism”, respectively (Table S11).These results suggest that the functions of the GWAS-identified genes were similar to those of the DEGs, but the latter involved more genes in a wider range of biological processes.The genes associated with carbohydrate metabolism fell into 15 clear functional subcategories, whereas only four GWAS-identified genes were associated with carbohydrate metabolism (Fig.4B).
The GWAS-identified genes for STI and RCL were checked to the transcriptome-identified genes, 51 DEGs were the same as those identified from GASM-RTM-GWAS in the AC-RIL population,including 28 STI and 26 RCL genes(3 shared).These genes involved in functional categories I–VI were detected, accounting for 10.41%PV and 18.29%PV of STI and RCL,respectively(Table S9).Thus,the 51 GWAS genes identified by forward genetics were confirmed using the transcribed genes identified by reverse genetics.
Transcriptome analysis may have spatial/time constraints, and gene expression may vary over time.A subsidiary experiment with additional transcriptome analysis of the multiple stem tip specimen on day 40 was performed with a single replicate for verifying the day 30 results and detecting differential DEGs by sampling stage.(For its relative consistency among replicates of transcriptome analysis, please refer to those of the day 30 in Table S10,accordingly, transcriptome data on day 40 may be considered as good approximations.) Assembly of mapped reads resulted in the identification of 32,631 genes in all samples on day 40 (Fig.S2C).Four pairs of comparisons, AT40vs.ACK40, ACK40vs.CCK40, AT40vs.CT40, and CT40vs.CCK40revealed 9276, 5982,11,096,and 9146 significant DEGs, respectively (Fig.S3B).Comparing the transcriptomic data between days 30 and 40 (Fig.S3C), the DEGs of the same parent differed between the two stages, with only a part shared.On days 30 and 40, 11,771 and 20,110 DEGs were respectively identified,in a total of 21,475 DEGs,with 10,406 ones shared(Fig.S3C).Using the same calculation for day 30, in the Venn diagrams on day 40, 16,021 DEGs (2503 + 743 + 544 + 2474 + 898 +645 + 2350 + 2241 + 1091 + 2532) were responsible for shading stress (Fig.4A).A set of 72 GWAS-identified genes (39 STI + 36 RCL-3 shared) were differentially expressed on day 40, of which 29 (17 STI + 14 RCL-2 shared) were the same as those on day 30(Fig.4A; Tables S6, S7).Thus, 94 (51 + 72-29) of 277 GWASidentified genes were identified by the two times of transcriptome analyses(Fig.4A;Tables S6,S7).This set of 94 DEG-verified GWAS genes were distributed in the complete set of six functional categories (Table S9).Since the 94 DEGs identified and expressed on day 30 and day 40 specimens separately, it indicates the GWASidentified gene may play roles at different stages.
3.5.Key shade-tolerance genes in the forward and reverse genetic system
The GASM-RTM-GWAS integrated with transcriptome analysis identified the shade-tolerance gene system, in which at least 277 genes with multiple biological functions were involved.To understand the shade-tolerance gene network, two approaches can be considered: to start from individual function categories or to start with a key gene or genes.For the latter,the genes(i)shared by both STI and RCL, (ii) identified by GASM-RTM-GWAS verified as DEGs,and(iii)as a hub with largest links to others in PPI were considered as key genes,from which five were selected(Table 2).In this study,
gST03.04/gRCL03.03 (Glyma.03G011200), gST17.01/gRCL17.01 (Glyma.17G127700), and gST18.12/gRCL18.09 (Glyma.18G078600) were the most likely key genes, given that they were detected for both STI and RCL as DEGs.The gST03.04/gRCL03.03 encodes AT-hook motif DNA-binding family protein, closely associated with abiotic stress[36,37]; the gST17.01/gRCL17.01 is with an unknown biological pathway in category VI;the gST18.12/gRCL18.09 participates in Fe2+/Zn2+transmembrane transporter activity in category II,closely associated with plant photosynthesis [38,39].The next group of key genes were gST15.05 (Glyma.15G156900) and gRCL15.13 (Glyma.15G172100), attractive as a hub with broadest links to others(79 and 95 genes,respectively),with functions involving hydrolase activity and DNA-directed RNA polymerase activity, respectively,in category V (Table 2).
4.Discussion
4.1.Advantages and features of GASM-RTM-GWAS for bi-parental RIL population compared to other procedures
This study found that GASM-RTM-GWAS has five advantages and features used for bi-parental RIL population.First, GASMRTM-GWAS detected more less-contribution genes than CIM.A high-density genetic map of the AC-RIL population containing 2253 GASMs was constructed(Table S12).In each confidence interval in CIM, there is a gene that overlapped with GASM-RTMGWAS-identified gene (Table S13), including the largestcontribution genes of STI and RCL.However, GASM-RTM-GWAS identified genes composed of large-contribution and many more small-contribution genes,with their probabilities and allele effects provided (Tables S4, S5, S6, and S7).The difference between heritability value and total gene contribution was calculated as the collective contribution of minor genes or polygenes which could not be obtained by CIM,indicating the higher detection power and efficiency of the GASM-RTM-GWAS procedure.Second, GASM-RTMGWAS used for bi-parental RIL population was also more powerful and effective than SNPLDP-RTM-GWAS,even though the same statistical procedure was used, but the marker types were different.The latter identified 122 STI and 140 RCL QTL(Fig.S4)and required an indirect inference step for genes,which was not the case for the GASM-RTM-GWAS.Third, the STI and RCL gene-allele matrices established from the GASM-RTM-GWAS provided a compact form of the relatively full genetic information of the population, from which various types of genetic information about the gene, allele effects, allele constitution of the RILs, recombination potential,and optimal genotype could be obtained or predicted.In addition,the recombination/breeding potential was high enough estimated from the gene-allele matrix,therefore,the optimal cross/genotypes can be designed for shade-tolerance among the RILs between Aijiaozao and Chippewa.Fourth, the interaction between 99 STI and 101 RCL genes was connected to the PPI networks, indicating that GASM-RTM-GWAS can effectively detect the gene system regulating shade-tolerance from different indicators, implying that shade-tolerance is a complex trait involving genes from two even more indicators and multiple biological processes(Fig.3B).Among them, key hub genes may be the major genes and may provide important information for a comprehensive study of shadetolerance.Fifth, the 51 genes or 94 genes identified by the GASM-RTM-GWAS were verified by transcriptomic analysis on day 30 and day 30+40, respectively, indicating that the genes detected by association analysis but in fact expressed at different growth stages.Furthermore, the GWAS-detected genes, including those in PPI networks,and the transcriptome-identified DEGs were both involved in functional categories I–VI, further showing that the genes detected by GASM-RTM-GWAS were relatively reliable,implying that the survival of plants under stress depends on therecognition of environmental stimuli, signal generation and transmission, gene expression, and metabolic regulation [40,41].

Table 2 Key STI and RCL genes in the AC-RIL population.
4.2.Advantages and features of integrating forward with reverse genetic analysis in understanding the gene network of shade-tolerance
The advantages and features of integrating forward with reverse genetic analysis are summarized as follows.(i) Trait gene expression is a chronological/spatial process, with many more DEGs involved in the expression processes from time to time.The expression of some causal genes involving many genes in a series of functional processes, including physiological, developmental, and cell physical and chemical functions.(ii) In only two time points,the 94 out of 277 GWAS-identified genes were verified by reverse genetic analysis, we expect that all GWAS-detected genes could be verified if more time points added.(iii) The above fact of GWAS-identified gene expressed at differential growth stages was found through the integrated forward and reverse genetic analysis, therefore, the shade-tolerance genes expressed neither starting from the very beginning nor working through all the time.(iv) Transcriptome analysis identified DEGs many more than those from GASM-RTM-GWAS (21,475 vs.277), the large difference between the forward and reverse analyses invites further investigation.
DEGs in samples at different time points were different not only in counts, but also in expression levels and directions (upregulated vs.downregulated).This study was straightforward in that all significant DEGs regardless of expression level and direction were treated as an identical DEG.This approach was used to count DEGs and not necessarily for gene function studies.Some DEGs shared between the comparisons of AT30vs.ACK30and CT30vs.CCK30were expressed in different directions.Light is considered a major threat to legume growth under shade stress [42–45], and photosynthesis-associated metabolic pathways (including ko00195, ko00196, and ko00710)belong to the subcategory of Energy Metabolism in KEGG (Fig.4B).In AT30vs.ACK30and CT30vs.CCK30(Table S14), respectively 15 and 3 DEGs (one shared DEG) of the photosynthesis pathway(ko00195) were identified.In the photosynthesis-antenna protein pathway (ko00196), the expression of DEGs was opposite between the two comparisons, with three DEGs upregulated for AT30vs.ACK30and five downregulated for CT30vs.CCK30.Thus, the upregulation of photosynthesis and photosynthesis-antenna protein genes may increase the efficiency of photosynthesis, and long-term shading treatment might also induce shade-tolerance regulation in sensitive materials.In the carbon fixation of the photosynthetic organism pathway (ko00710), AT30vs.ACK30had 10 upregulated DEGs,whereas CT30vs.CCK30had 15 upregulated DEGs,in agreement with previous reports on the effective utilization of carbohydrates[46,47].In three KEGG pathways, the distribution of DEGs on day 40 was similar to that on day 30 (Table S14).Respectively 10 and 8 shared DEGs in the two stages showed different expression levels and directions between the two comparison sets.The differences in gene expression levels and directions between days 30 and 40 may be due to allele differences or allele epistasis.
4.3.Probing into STI and RCL as well as the shade-tolerance gene system
STI and RCL are closely related because of an internal relationship in which cell length is the basis of plant height, whereas RCL is relatively stable and less affected by the environment [48].This could explain why they had a similar set of six functional gene categories but with different contents.STI is suitable for quick and large-scale evaluation, especially in breeding programs, whereas RCL is more suitable for studying the shade-tolerance mechanism,especially cell and tissue mechanisms.Nine genes associated with cell wall traits were detected in RCL,but not in STI.Plant cell walls not only regulate the size and shape of plant cells providing structural support but also produce complex dynamic changes in response to developmental and environmental signals to play a defensive role under biotic and abiotic stresses [49–52].Thus,exploring the shade-tolerance gene system from the external STI to the internal RCL has provided more complete information for understanding the components of the shade-tolerance gene system.We speculated to study shade-tolerance full mechanism, a combination of morphological, anatomical, physiological and biochemical indicators should be considered.
The STI and RCL gene systems composed of 277 genes were detected from the association between their phenotype and GASM,whereas 21,475 DEGs were identified from transcriptome under shading vs.non-shading conditions.In effect, the latter represents a whole gene system conferring shade-tolerance, whereas the former represents the gene system of the two specific indicators STI and RCL.This may explain the large differences between the two systems (GWAS vs.RNA-seq).Of course, the difference between forward and reverse genetic analyses is a major reason.However,the response of soybeans to shade stress involves multiple morphological, anatomical, physiological, and biochemical traits from the seedling to the adult stage, which have not been studied yet,except for STI and RCL in the present study [11,53–58].This can explain the many DEGs (21, 475) detected in shading/nonshading transcriptomic analysis but relatively few genes (277)identified in GWAS for the two indicators STL and RCL [59].That is,the transcriptome is a collection of gene expression at the overall level of the tested sample, reflecting almost all the changes in traits due to shading stress and providing information on gene expression systems far greater than GWAS analysis.Comparison results of GWAS and transcriptome analysis further confirms the importance of integrating forward and reverse genetic analyses to elucidate the entire genetic system of a trait.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statement
Yanzhu Su:Conceptualization, Methodology, Investigation, Data curation, Writing – original draft.Xiaoshuai Hao:Investigation.Weiying Zeng:Investigation, Resources.Zhenguang Lai:Investigation, Resources.Yongpeng Pan:Investigation, Resources.Can Wang:Methodology.Pengfei Guo:Methodology, Validation.Zhipeng Zhang:Investigation.Jianbo He:Methodology, Validation.Guangnan Xing:Supervision, Project administration.Wubin Wang:Supervision, Project administration.Jiaoping Zhang:Supervision, Project administration.Zudong Sun:Investigation,Formal analysis, Resources, Supervision, Project administration,Funding acquisition.Junyi Gai:Conceptualization, Methodology,Visualization, Formal analysis, Resources, Supervision, Project administration, Funding acquisition, Writing – review & editing.
Acknowledgments
This work was financially supported by the grants from the National Key Research and Development Program of China(2021YFF1001204, 2021YFD1201602), the MOE 111 Project(B08025), the MOA CARS-04 program,the Program of Jiangsu province (JBGS-2021-014), the Guangxi Scientific Research and Technology Development Plan (14125008-2-16), and the Guidance Foundation of Sanya Institute of Nanjing Agricultural University(NAUSY-ZZ02, NAUSY-MS05).The funders had no role in work design, data collection and analysis, and decision and preparation of the manuscript.We thank the Sugarcane Research Institute,Guangxi Academy of Agricultural Sciences for providing the light microscope.
Appendix A.Supplementary data
Supplementary data for this article can be found online at https://doi.org/10.1016/j.cj.2023.11.013.

- The Crop Journal的其它文章
- Flowering-time regulation by the circadian clock: From Arabidopsis to crops
- Global characterization of OsPIP aquaporins reveals that the H2O2 transporter OsPIP2;6 increases resistance to rice blast
- Drought-triggered repression of miR166 promotes drought tolerance in soybean
- The OsBSK1-2-MAPK module regulates blast resistance in rice
- Natural variation of an autophagy-family gene among rice subspecies affects grain size and weight
- Rice gene OsUGT75A regulates seedling emergence under deep-sowing conditions