
A telomere-to-telomere genome assembly of Zhonghuang 13,a widely-grown soybean variety from the original center of Glycine max
2024-03-07 01:49AnqiZhngTnghoKongBiqunSunShizhengQiuJiheGuoShuyongRunYuGuoJiruiGuoZhishuiZhngYueLiuZhengHuToJingYdongLiuShuqiCoShiSunTingtingWuHuilongHongBingjunJingMoxingYngXingyuYoYngHuBoLiuTinfuHnYdongWng
The Crop Journal 2024年1期
Anqi Zhng, Tngho Kong, Biqun Sun, Shizheng Qiu, Jihe Guo, Shuyong Run, Yu Guo,Jirui Guo, Zhishui Zhng, Yue Liu, Zheng Hu, To Jing, Ydong Liu, Shuqi Co, Shi Sun,Tingting Wu, Huilong Hong, Bingjun Jing, Moxing Yng, Xingyu Yo, Yng Hu,*, Bo Liu,*,Tinfu Hn,*, Ydong Wng,*
a Center for Bioinformatics, School of Computer Science and Technology, Harbin Institute of Technology, Harbin 150001, Heilongjiang, China
b Ministry of Agriculture Key Laboratory of Soybean Biology (Beijing), Institute of Crop Sciences, Chinese Academy of Agricultural Sciences, Beijing 100081, China
c National Key Facility for Crop Gene Resources and Genetic Improvement, Institute of Crop Sciences, Chinese Academy of Agricultural Sciences, Beijing 100081, China
ABSTRACT
1.Introduction
Soybean (Glycine max [L.] Merr.), originating in China, hold a paramount position as one of the most crucial oil and protein crops.They contribute to more than a quarter of the protein utilized in both food and animal feed[1–5].It is widely acknowledged that the cultivated soybean emerged through the domestication of its wild annual progenitor, Glycine soja, around 5000 years ago from the Yellow River Basin in temperate regions of China.This specific geographical range represents the greatest allelic diversity of soybean [6,7].Subsequently, its distribution expanded northward to encompass high-latitude cold zones and southward to encompass low-latitude tropical regions.Therefore, the exploration of genetic resources within the origin region bears immense significance in advancing the global frontiers of soybean breeding.
Zhonghuang 13 (ZH13), a soybean cultivar meticulously developed and released by Chinese breeders in 2001, occupied the largest planting area in the first two decades of 21st century in China, and stood as a testament to advanced agronomic traits and remarkable adaptability to wide regions including Yellow River Basin, southern Northeast, and some parts of Northwest and South China[8,9].In comparison to the widely recognized Williams 82 cultivar, ZH13 boasts heightened genetic diversity and ecological type of origin reign [10].Furthermore, ZH13 is an ideal variety in the breeding strategy called ‘‘Potalaization”, which allows breeding of novel widely adapted soybean varieties through the use of multiple molecular tools in existing elite widely adapted varieties [7].
Whole-genome sequencing of ZH13 has been previously conducted [8,9].This approach enables the identification of crucial genes and genetic variants linked to favorable traits, thereby enhancing our comprehension of soybean breeding [5,11–16].Nonetheless, limitations inherent in second-generation sequencing, including inadequate coverage of the genome and challenges in precisely assembling and annotating repetitive genomic regions,such as telomeres and centromeres, have resulted in the persistence of over 1000 gaps within the most recent soybean reference genome [17–21].
Telomere-to-Telomere (T2T) assembly is a state-of-the-art genomic sequencing method that employs long-read sequencing platforms such as Pacific Biosciences (PacBio) or Oxford Nanopore Technologies (ONT) to obtain the comprehensive sequence from one telomere to another, encompassing highly repetitive regions,centromeres, and telomeres [22–24].This approach yields a complete and contiguous assembly of the entire genome [23,25].T2T assembly effectively overcomes the limitations associated with sequence gaps and assembly errors that are frequently encountered in whole-genome sequencing [26].Moreover, it offers enhanced resolution for the detection and characterization of large-scale SVs [27,28].Consequently, T2T assembly exhibits immense potential in expanding our knowledge of intricate genomes, such as soybeans, and driving advancements in breeding programs.
In this study, we utilized T2T assembly to conduct de novo assembly of ZH13 genomes.By employing this innovative genomic assembly approach, we aim to deliver a fully covered soybean sequence encompassing 100% of the genome.This significant advancement will enhance our comprehension of the structure and functional significance of the soybean genome, and also provide a reference genome sequence for elite cultivar improvement.
2.Materials and methods
2.1.Plant material preparation and genome sequencing
The soybean seeds of Glycine max,cv.ZH13 were from the Institute of Crop Sciences, Chinese Academy of Agricultural Sciences.Four soybean seeds were planted in 10 L pots on June 5, 2023,and grown outdoors under natural conditions in Beijing, China(39.95°N, 116.32°E).On Day 20 after emergence (VE), the fresh young leaf tissue was collected and frozen immediately in liquid nitrogen for DNA extraction.The High Molecular Weight (HMW)DNA extraction was performed using the modified cetyltrimethylammonium bromide (CTAB) method and large fragments(>100 kb)were separated using the SageHLS HMW library system.The standard libraries were constructed for subsequent sequencing.For PacBio HiFi sequencing, the library was constructed using SMRTbell Express Template Prep Kit.For ONT ultra-long sequencing, the library was created using SQK-ULK001 kit.For WGS sequencing, the library was created using NEBNext Ultra II DNA Library Prep Kit.For Hi-C sequencing, fresh leaves were fixed in 4% (v/v) formaldehyde after grinding with liquid nitrogen.Cell lysis, chromatin capture and digestion, and DNA quality check were performed according to the modified methods from [29].The library was created using NEBNext Ultra II DNA Library Prep Kit.A PacBio Revio sequencer was used to produce a 96.89 Gbp Hifi datasets (mean read length: 100.7 kb) and a PromethION 48 sequencer was used to produce a 96.63 Gb ultralong dataset(mean read length: 100.7 kb).Moreover, the 150 bp pair-end WGS(55.40 Gb)and Hi-C(106.4 Gb)datasets were produced by an Illumina Novaseq 6000 sequencer.
2.2.Draft genome assembly by PacBio Hifi and ONT ultralong reads
The PacBio Hifi reads were input to Hifiasm [30] (version:0.19.5-r590, default parameters) to generate a Hifi graph, and the ONT ultralong reads were then aligned to the graph to produce choromosome-level contigs [31].For NextDenovo [32] (version:2.5.2, parameters: read_cutoff = 1 k, genome_size = 1 g), the ONT ultralong reads were input at first to produce initial contigs which were further polished by NextPolish [33] (version: 1.4.1, parameters: -x map-hifi -min_read_len 1 k -max_depth 100) with input PacBio Hifi reads.For Canu [34] (version: 2.2, parameters:genomeSize=1 g),only the PacBio Hifi reads were input to produce Hifi-only contigs.
The long (> 1 Mb) Hifiasm contigs were employed as primary contigs at first.Moreover, BLAST was employed to check the 851 < 1 Mb Hifiasm contigs.We found out that 759, 56 and 30 of the short contigs can be aligned to chloroplast, mitochondrion and rDNA, and others can also be aligned to repeats of soybean genomes as well.Thus,they were filtered out and the primary contig set did not update since we would like to build a concise draft assembly and solve potential gaps and mis-assemblies with the supplement of other assemblers.The primary contigs were then aligned to the current version of ZH13 reference (termed as Gmax_ZH13_v2.0) by minimap2 [9,35] (version: 2.26-r1175,parameters: -x asm5 -f 0.02) to determine their orders and orientations.A draft ZH13 assembly was then generated and all the Pac-Bio reads, ONT reads, NextDenovo contigs and Canu contigs were aligned to it by minimap2 (parameters: -x map-pb -r 1000; -x map-hifi-r 1000;-x map-ont-r 10000;-x asm5-f 0.02)for further processing.
2.3.Refinement of draft assembly
An in-house script was used to divide the draft assembly into 10 kb sliding windows and scan the read coverages to detect HCRs and LCRs.Herein,an HCR(LCR)is defined as a window whose coverage is > 200 (< 30).The local sequences of HCRs and LCRs were then searched by BLAST to check their homologies as an evidence of mis-assembly or not.Moreover, the numbers and positions of read clippings were also investigated as they are more important signatures to discover mis-assemblies.Since the HCR sequences can be aligned to mitochondria,chloroplast,mRNAs or mobile elements,their high coverages could be not due to mis-assembly,but plausibly the affection of the reads from those elements as well as the aligner’s own strategy to handle repetitive reads.So,they were not considered for correction.
The two LCRs and four remaining gaps in the draft assembly were then reconstructed in two steps.Firstly, we collected the NextDenovo and Canu contigs which can span those regions.The local sequences were then replaced by corresponding contigs with the guidance of nearby anchors.The reads were also re-aligned after the reconstruction to re-check local coverage.Moreover, we selected the contig leading to a local coverage closest to the mean coverage of the whole genome, if multiple candidates exist.It is worth noting that this approach can either solve the LCRs/gaps or turn them to HCRs, since the employed contigs can at least reflect most of the elements existed in local regions,even if the copy numbers are incorrect and/or some of the local sequences are still absent.
For the still unsolved HCRs,we used an in-house tool to implement local assembly.Given an HCR, the tool collected the reads harbored or anchored to that region at first (termed as active reads) and iteratively assembles them.In each iteration, the tool separately tries each of the active reads to extend the local sequence from the region boundary, and aligns other reads to the extended sequence (by BLAST).If there are enough reads being aligned with high scores, the HCR is updated and a number (relative to the mean read coverage of the whole genome) of highest scored reads are removed.The procedure continues until the contig reaches the other boundary of the HCR, or no active read remains.The produced contig is then integrated into the genome with manual curation and read coverage checking.
2.4.Telomere identification and refinement
The 7-mer repeats(CCCTAAA/TTTAGGG)were used to identify telomeres in the draft assembly.37 telomeres from 2212 to 18154 bp in length were identified.Further, we used the 7-mer motif to search the contigs produced by NextDenovo, Canu and Hifiasm (using hifi reads only), and identified the three missing ones.Two (Gm10 and Gm15) were supplied by Canu and one(Gm09) was supplied by Hifiasm.Moreover, it was found that the Gm02 upstream telomere produced by Canu (8449 bp) was obviously longer than that of Hifiasm (3045 bp), so that we updated it.The precise locations of telomeres within the ZH13-T2T genome were ascertained by using seqtk (https://github.-com/lh3/seqtk).The command used was ‘seqtk telo -s 1 -m CCCTAAA ref.fa’.
2.5.Genome-wide comparisons and identification of SVs
We conducted a comparative analysis using publicly available ZH13 soybean genome data and the assembled T2T assembly results.First,we aligned the ZH13-T2T genome data to the soybean reference genome using Minimap2 (Version 2.26-r1175) (https://github.com/lh3/minimap2) [35,36].Minimap2 was utilized to map the long sequencing fragments, present in fastq format files of each sample, to the reference genome provided in fasta format[35].To enhance comparative efficiency, we utilized the ‘‘-ax asm5 –eqx” parameters for fragment alignment, set the software to work with a maximum of 96 threads using the ‘‘-t” parameter,filtered out regions with sequence differences greater than 5%,and stored the results of sequence matches or mismatches in SAM format.All other comparison parameters during the process were left at their default settings.We subjected the comparison results of the two genome versions to structural variation detection using the SyRI mutation detection tool (https://github.com/schneebergerlab/syri), configured with default parameters, and saved the mutation detection results as a ‘‘syri.out” file [37].Subsequently,we employed the Plotsr software to visualize the mutation detection results, using default parameters for the transformation process, and saved the generated images in PDF format [38].The visualization command used was ‘‘plotsr --sr syri.out --genomes genome.txt”.
2.6.Identification of rDNA and non-coding RNA
5S rRNA is transcribed from the 5S DNA,while 48S rRNA is composed of 28S rRNA, 5.8S rRNA, and 18S rRNA.We identified tRNAs using tRNAscan-SE v2.0,rRNAs using Barrnap v0.9(https://github.-com/tseemann/barrnap), and miRNA and snRNA using INFERNAL(https://eddylab.org/infernal/) against the Rfam (release 12.0)database [39–41].Copy numbers of both 5S rDNA and 48S rDNA were determined using Barrnap.Complete copies of 5S rDNA and 48S rDNA were used as input for genotype identification.Subsequently,a multiple sequence alignment was performed on 5S rDNA and 48S rDNA using MAFFT with default parameters(https://mafft.cbrc.jp/alignment/software/) [42,43].For the 5S rDNA and 48S rDNA, genotype analysis was conducted using single nucleotide polymorphisms (SNPs) and insertions/deletions (InDels)with over 10% support from the 5S rDNA copies.It is important to note that all selected indices for 48S rRNAs were located within intergenic spacer regions.
2.7.Repeat identification and gene annotation
We utilized the EDTA pipeline (https://github.com/oushujun/EDTA) for transposable element (TE) annotation [44,45].The main steps involved in identifying repetitive sequences in the genome were as follows: First, we created an indexed database using the RMBlast engine.Then, we employed RepeatModeler (https://www.repeatmasker.org/RepeatModeler/) for de novo prediction,which involved five iterative rounds to obtain repetitive sequences and Stockholm format seed alignment files[46].Subsequently,we performed genome annotation using RepeatMasker [47–49].Lowcomplexity sequences and small RNA (pseudo) genes were not masked, and the search for insertions of missing sequences was disabled.Repeat-masked genome and repeat sequence library constructed by RepeatModeler and RepeatMasker were used for subsequent TE analysis.
For ab initio annotation, we utilized BUSCO (https://github.-com/metashot/busco) to create a training dataset for Augustus(https://github.com/Gaius-Augustus/Augustus) [50].Based on this training dataset,we further applied Augustus to predict the coding regions of genes on the masked genome.We further analyzed the component composition of the previous gap region and enriched the new annotated genes by GO and KEGG using KOBAS, with P < 0.05 as the threshold [51].To analyze the gene expression in the gap regions,we obtained RNA-seq data of Glycine max and Glycine soja from the study by Liu et al.[1].The tissue we used were V1 stage root(A),V1 stage stem(B),V1 stage leaf(C)and R1 stage leaf (D).We analyzed the RNA-seq data using the genomic and annotation files from our study, with a threshold of TPM > 1 for gene expression.The detailed information of the selected samples can be found in Table S1.
2.8.Centromere localization
We employed Tandem Repeat Finder (TRF, version 4.09.1)(https://github.com/Benson-Genomics-Lab/TRF) to discern and classify satellite, small satellite, and microsatellite sequences within the soybean T2T genome [52].The default parameters utilized for TRF were set to ‘2 7 7 80 10 50 500 -f -d -m’, and the results of TRF annotation were merged using TRF2GFF (https://github.com/Adamtaranto/TRF2GFF).We manually eliminated tandem repeats with fewer than five copies and redundant occurrences.Sequences characterized by lengths of less than 10 bp,between 10 bp and 100 bp, and exceeding 100 bp were respectively categorized as microsatellites, minisatellites, and satellites.Building upon the results obtained from the previous EDTA pipeline and TEsorter (https://github.com/zhangrengang/TEsorter), we obtained TE annotation files and the total number of copies of different period sequences in various chromosomes[44,45,53].Utilizing two high-copy satellite repeat subfamilies, CentGm-1 and CentGm-2, which are exclusive to the centromeric region, we ascertained the approximate location of the centromere [54].Lastly,by employing the Integrative Genomics Viewer(IGV)browser, we observed an overlap between the regions with TE annotation loss and the region where the 91/92 bp-long sequences were concentrated, thereby identifying the centromere region [55–60].
3.Results
3.1.T2T assembly of the soybean ZH13 genome
Four types of sequencing data were initially produced for a single ZH13 sample,including PacBio Hifi reads(96.89 Gb),ONT ultralong reads (96.63 Gb), Illumina whole genome sequencing (WGS)(55.40 Gb) and Illumina high-throughput chromosome conformation capture (Hi-C) reads (106.4 Gb).We only used the long reads(PacBio Hifi and ONT ultralong) to implement T2T assembly, and the short WGS and Hi-C reads were employed to assess assembly quality.The assembly was implemented by a pipeline based on multiple assemblers and in-house tools in three phases as following (a flowchart is in Fig.1).
1) Draft assembly.At first, three set of contigs were independently produced by various assemblers, i.e., Hifiasm [30,31],NextDenovo [32] and Canu [34].Both of Hifiasm and NextDenovo used all the PacBio Hifi and ONT ultralong reads, and Canu used PacBio reads only.The 23>1 Mb contigs produced by Hifiasm were employed as primary contigs and aligned to Gmax_ZH13_v2.0 by minimap2[9,35].22 of them can be colinearly aligned and 1 contig were aligned to two different chromosomes.We manually checked this split alignment and confirmed that it was a mis-assembly caused by Hifiasm.The contig was then divided into two.A 24-contigs draft assembly was then generated, which 17 of the 20 ZH13 chromosomes were covered by a single contig,2 and 1 chromosomes have 2 and 3 contigs,respectively.The PacBio reads,ONT reads and the contigs produced by NextDenovo and Canu were aligned to the draft assembly for further refinement.
2)Assembly refinement.There were four remaining gaps in the draft assembly.Moreover,we also detected high-and low coverage regions(HCRs and LCRs)by an in-house script as they could be also mis-assembled regions.43 HCRs and 2 LCRs were found.We searched the sequences of the HCRs by BLAST and the results indicated that all of them can be aligned to mitochondria, chloroplast,mRNAs or mobile elements.Thus, we realized that they could be not mis-assembly.However, plenty of read clippings were observed around the two LCRs, which indicated mis-assembles.Thus, the four gaps (gap1: CM010418.2: 18,024,780–18,025,280,gap2: CM010419.2:27,778,852–27,779,352, gap3: CM010421.2:3,326,824–3,327,324 and gap4: CM010421.2: 40,056,171–40,056,671) and the two LCRs (LCR1: CM010409.2: 15,403,000–15,404,000 and LCR2: CM010427.2: 15,777,563–16,073,378) were refined with spanning NextDenovo and Canu contigs (refer to Methods for more detailed information).Gap2, gap4 and LCR1 were successfully reconstructed by two NextDenovo contigs and one Canu contig (Figs.S1-S3), respectively.However, Gap1, gap3 and LCR2 were turned to be HCRs (Figs.2, 3, S4), indicating that the spanning contigs also cannot well-handle them.Highly repetitive sequences were found there,i.e.,gap1 is full of LTR retrotransposons,while gap3 and LCR2 are rDNA arrays.Further,an in-house local assembly tool was employed to iteratively collect and tile the reads anchored to the corresponding regions to refine the assembly.The solved Gap1 is 467.3 kb long which mostly consists of gypsy and copia.Gap3 and LCR2 are about 4.15 Mb and 414 kb long, respectively, having 545 48S rDNA copies and 1269 5S rDNA copies.We manually checked the read re-alignments to the three regions with IGV [56] and normal coverages were observed(Figs.2, 3, S4).The genomic structures of all gaps were presented in the Fig.S5.
3) Telomere identification and refinement.37 telomeres were identified from 17 chromosomes of the draft assembly.We further checked the contigs produced by NextDenovo, Canu and Hifiasm(using Hifi reads only),and reconstructed the three missing telomeres,i.e.,Gm09(downstream,3831 bp),Gm10(upstream,5889 bp)and Gm15(downstream,9764 bp).Thus,all the 40 telomeres were recovered with an 8449 bp median length.
Finally, a complete genome of ZH13 (termed as ZH13-T2T,Fig.4) was generated whose total length is 1,015,024,879 bp (no gap, N50: 52,033,905 bp).The quality of the assembly was evaluated by various metrics and four issues are observed as following.Firstly, the complete BUSCO metric [61] (99.8%, lineage dataset:embryophyta_odb10)suggests its high completeness.More importantly, all the 393 gaps of Gmax_ZH13_v2.0 have been filled.Secondly, Illumina WGS read-based Merqury’s Qv metric [62]reaches 46.441, suggesting that it also achieves high base-level accuracy.Thirdly,it is observed from the Hi-C map(Fig.S6,generated by Juicerbox [63]) that strong interactions are concentrated along the diagonal, indicating that no obvious mis-assembly can be discovered from the view of Hi-C data.Fourthly, with careful detection and correction of HCRs and LCRs,the coverages of PacBio Hifi and ONT Ultralong reads are nearly uniform along the whole ZH13-T2T genome,also suggesting that the assembly could be free of mis-assembly.
3.2.Genome-wide comparison to Gmax_ZH13_v2.0 and other soybean T2T assemblies
We compared ZH13-T2T with Gmax_ZH13_v2.0 by SyRI [37](Fig.4).Most of the ZH13-T2T chromosomes of are longer, mainly due to the filled gaps.There are also 421>5 kb not-aligned regions(NARs, 16.3 Mb in total), indicating that the corresponding local sequences are quite different.Meanwhile, SyRI also identified 112 structure variations(SVs),i.e.,30 inversions,15 translocations and 67 duplications.Most of them are in the NARs and highly complex,i.e.,the combinations of multiple inversions and duplications.
We further aligned the ONT ultralong reads of ZH13-T2T to Gmax_ZH13_v2.0(by minimap2).The local alignments in the NARs showed concentrated and extremely complex SV signatures, i.e.,plenty of large clippings, split alignments and abnormal local coverages,especially for those SV-surrounding regions(an example is in Fig.S7).On the contrary, colinear alignments with normal coverage were observed from the corresponding regions of ZH13-T2T, suggesting no obvious SV signature there.We also investigated the alignments on the NARs without SVs and similar results were observed (Fig.S8).Under such circumstance, we realize that although the different donor samples potentially have divergences in genomic sequences, there could be also a number of misassemblies in Gmax_ZH13_v2.0, possibly due to the limitation of its sequencing data.Moreover, considering the consecutive read alignments on ZH13-T2T,the mis-assemblies should have been largely resolved.
We further compared ZH13-T2T to two newly published T2T soybean genome assemblies, one is from Wm82 [64] (produced by Northeast Agricultural University, termed as Wm82-NJAU)and the other is also from ZH13 (produced by Guangxi University and Northeast Agricultural University, termed as ZH13-T2T-GN)[65].
For the comparison between ZH13-T2T and Wm82-NJAU, 167 NARs (23.02 Mb in total) and 30 SVs (16 inversions, 7 translocations and 7 duplications) were identified.To investigate the NARs and SVs, we also downloaded the PacBio Hifi and ONT ultralong datasets of Wm82-NJAU and re-aligned them to the genome.In a large proportion of the investigated regions, both of ZH13-T2T and Wm82-NJAU have normal read alignments (Fig.S9), suggesting the differences between the two soybean genomes.However,in some of the NARs, abnormal read alignments can still be found from Wm82-NJAU but not for ZH13-T2T (Fig.S10).Moreover, we also checked the local read coverages along the whole Wm82-NJAU genome and found 12 HCRs and 107 LCRs.
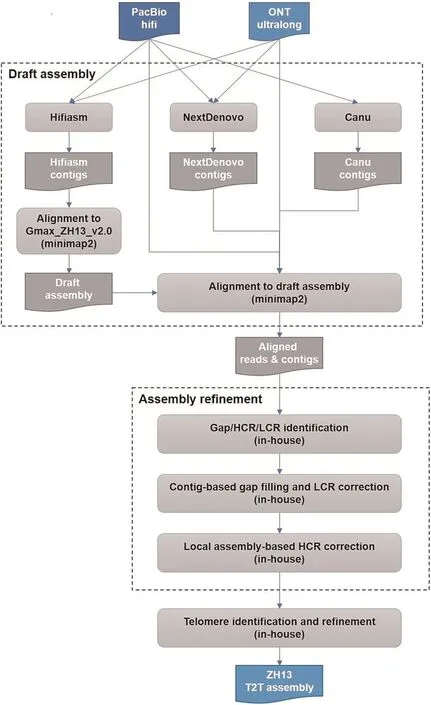
Fig.1.ZH13-T2T assembly pipeline.
For the comparison between ZH13-T2T and ZH13-T2T-GN,high consistency was observed (Fig.S11), i.e., most of the genomic regions coincide with each other.However,there were still 4 NARs and 69 SVs (4 inversions, 65 duplications) identified.We investigated the read re-alignment results in the inconsistent regions,and found abnormal alignments and coverages for ZH13-T2T-GN,but not for ZH13-T2T.One example is given in Fig.S12 that a relatively high coverage (i.e., HCR) was observed, indicating that the copy number of local tandem repeats could be reduced.A more obvious example is in Fig.S13 that the assembled long 48S rDNA array (i.e., gap3) of ZH13-T2T is collapsed in ZH13-T2T-GN.Only 130 kb rDNA region was assembled (comparing to 4.16 Mb of ZH13-T2T) and an HCR with extremely high coverage was observed there.Moreover, two additional issues were also observed.One is that telomere motifs were not identified for four chromosome-ends of ZH13-T2T-GN (i.e., GWHBWDJ00000003.1 upstream, GWHBWDJ00000004.1 downstream, GWHBWD0000 0006.1 downstream and GWHBWDJ00000008.1 downstream).Another one is that there are additionally 12 HCRs and 33 LCRs in ZH13-T2T-GN besides the NARs (an LCR example is in Fig.S14).
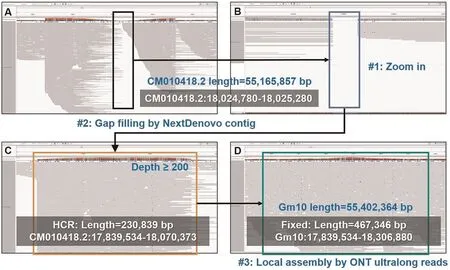
Fig.2.The filling of gap1.(A)the IGV snapshot of ONT ultralong read alignment in the surrounding region of gap1 in the draft assembly of ZH13.(B)a zoom-in view of gap1.(C) the IGV snapshot of ONT ultralong read alignment after NextDenovo contig correction (HCR was observed).(D) the IGV snapshot of ONT ultralong read alignment after local assembly-based correction with anchored ONT ultralong reads (consecutive alignments and normal coverage were observed).

Fig.3.The filling of gap3.(A)The IGV snapshot of ONT ultralong read alignment in the surrounding region of gap3 in the draft assembly of ZH13.(B)A zoom-in view of gap3.(C) The IGV snapshot of ONT ultralong read alignment after NextDenovo contig correction (HCR was observed and 48S rDNA array was identified).(D) The IGV snapshot of ONT ultralong read alignment after local assembly-based correction with anchored reads, consecutive read alignments were observed.Moreover,by manually checking the alignment details, we also found many reads having very low MAPQ, i.e., each of the reads had multiple candidate mapping positions and cannot be confidently aligned.Overall, normal coverage was proved by considering all the primary and secondary alignments of the reads.
3.3.Genome annotation and gene prediction
Complete T2T assembly of 20 chromosomes revealed that approximately 57.07% of the soybean genome consisted of annotated repeating elements.Among these elements,retrotransposons accounted for 38.16% (comprising 0.12% SINEs, 1.58% LINEs, and 36.47% LTR elements), while DNA transposons accounted for 6.72% (Table 1; Fig.S15; Supplementary data 1).Furthermore, we detected 3.64 Mb of microsatellites, 11.58 Mb of minisatellites,11.44 Mb of satellites, 0.41 Mb of 5S rDNAs, and 4.16 Mb of 48S rDNAs (Table S2).Collectively, these tandem repeats constitute 2.63% (26.65 Mb) of the soybean genome, which significantly surpasses the 1.03% (10.54 Mb) observed in the Gmax_ZH13_v2.0.Additionally, the intergenic spacer (IGS) region of 5S rDNA is approximately 220 bp in length, while the 5S region itself spans approximately 110 bp (Fig.5).There is a partial overlap region of around 153 bp between 5.8S rDNA and 28S rDNA.Based on the analysis of 1 InDels,the 48S rDNA has been categorized into 2 distinct genotypes.Furthermore, an examination of 13 SNPs and InDels has led to the classification of the 5S rDNA into 32 different genotypes.

Fig.4.The ZH13-T2T assembly and its comparison to Gmax_ZH13_v2.0 and Wm82-NJAU.The orange, blue and green lines indicate ZH13-T2T,Gmax_ZH13_v2.0 and Wm82-NJAU, respectively.Gray and blank blocks between various genomes indicate syntenic regions and NARs.Inversions,translocations and duplications are marked by filled orange, green and blue curves.
The annotation of the ZH13-T2T genome was performed using the Augustus software for ab initio annotation.After excluding transposon genes and applying a gene filtering process, a total of 50,564 high-confidence protein-coding genes were obtained(Supplementary data 2).Notably, in comparison to ZH13-2019,we identified 707 novel genes within the gap regions.The results of the Gene Ontology (GO) function and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis obtained through KOBAS (Knowledgebase for Ontology-based Functional Annotation and Analysis)reveal significant enrichments for newly discovered genes in various biological pathways.Specifically, the novel genes were involved in various biological processes, cellular components, and molecular functions, ranging from cell surface to nucleus,including negative regulation of transcription,DNA-templated(p=7.52E-05),protein autophosphorylation (p = 0.0003), positive regulation of transcription by RNA polymerase II (p = 0.0012), and mRNA export from nucleus(p = 0.0094) (Fig.S16).In the context of KEGG, these genes participated in phenylalanine, tyrosine and tryptophan biosynthesis(p=0.0004),fatty acid biosynthesis(p=0.0007),phosphatidylinositol signaling system (p = 0.0046), fatty acid metabolism(p = 0.0048), RNA transport (p = 0.0105), and the MAPK signaling pathway - plant (p = 0.0156) (Fig.S17).The RNA-seq analysis results indicate the presence of expression in a total of 295 genes across 38 gap regions (Tables S3-S6).The reason for not mapping all the gap regions and genes could be that some regions are not expressed or have extremely low expression levels.Nevertheless,our RNA-seq analysis results can serve as supporting evidence for these identified gap regions and genes.
In the previous gap regions,we observed the highest number of newly discovered genes within the 14.84–17.73 Mb region of chromosome Gm13,totaling 135 new genes.Furthermore,our analysis identified 42,668 TEs,300 GmCent-1 elements,and 586 GmCent-2 elements within these gap regions.Notably, the Gm12 chromosome’s 30.54–32.38 Mb region contained the highest count of TEs, with a total of 3416 TEs.In the Gm05 chromosome’s 20.40–24.24 Mb region, we observed the highest number of GmCent-1 elements, amounting to 131, and in the Gm11 chromosome’s 25.51–27.93 Mb region, the highest count of GmCent-2 elements,totaling 64 (Fig.S5).
3.4.Detection of centromere
The centromere,a crucial component of chromosome structure,consists of highly repetitive heterochromatin and plays a vital role in ensuring accurate chromosome segregation.In plants, the centromere region is characterized by an abundance of retrotransposon and tandem repeats [66].Investigating the potential functions of the centromere in genome evolution and chromatin assembly holds significant importance.However, current genome sequencing approaches face challenges in fully assembling the repetitive sequences within the centromere region.Here, we employed the TRF tool to identify repeat monomers within the ZH13-T2T genome that likely constitute the centromere.Consistent with previous studies, we found a large number of tandem repeats of 91 and 92 bp in length, complementing the gaps in TE of centromere region (Figs.6A–D, S18) [54].The heat map shows high similarity of sequences in the centromere region, indicating that the centromere region is highly tandem repetitive (Fig.6E)[59].

Table 1 Statistics of repetitive elements in ZH13-T2T.

Fig.5.Genome structure of 5S and 48S rDNA arrays.(A).Sequence structure of a typical 5S(up)and 48S rDNAs(down)repeat unit.IGS,intergenic spacer region;ITS,internal transcribed spacer region.(B).Variations of the most abundant genotypes of 5S rDNAs (up) and 48S rDNAs (down).(C).Genome structure of rDNAs (up) and 48S rDNAs(down).
Our findings indicate that the average length of the 20 centromeres analyzed is 2.40 Mb, with the longest centromere observed on Gm02 (4.42 Mb) and the shortest on Gm13(0.66 Mb) (Fig.6A).Notably, no significant correlation was observed between centromere length and chromosome size.Furthermore, the relative positions of centromeres varied among different chromosomes, with the minimum L/S ratio (long arm length/short arm length) recorded as 1.02 (Gm07) and the maximum L/S ratio as 2.95 (Gm15).A total of 8 genes were identified in the centromeric region of the soybean T2T genome.These genes were mainly enriched in chromatin DNA binding, mRNA cis splicing via spliceosome, histone binding, basal transcription factors,spliceosome, and pyrimidine metabolism (Fig.6F).

Fig.6.Genomic structure of telomere and centromere.(A) The length of the centromere.(B)The location of centromere in the genome.(C)The number of copies in the genome of tandem repeats with lengths of 91 and 92 bp.(D)The total length of tandem repeats of 91 and 92 bp in each chromosome.(E) The distribution and degree of correlation of various sequences in the centromere region are represented by heat maps.(F) Enrichment of genes in centromere region.
On average, centromere sequences were composed of 96.0%centromere satellite DNA (CentC), centromere retrotransposons(CRM), and other non-CRM Gypsy retrotransposons.The proportions of these components varied significantly across different centromeres, ranging from 0.0% to 73.3% for GmCent-1, 0.0% to 90.4%for GmCent-2, 0.0% to 2.2% for CRM, and 7.3% to 68.2% for other non-CRM Gypsy retrotransposons (Fig.S19).Almost all centromeres were rich in CentC, and there are no CentC-poor centromeres.
4.Discussion
Cultivated soybeans originated in China,it has undergone strict genetic bottlenecks during domestication, resulting in accessions from origin region possibly exhibiting high genetic diversity.In this context, the cultivar ZH13, developed in 2001 through meticulous breeding efforts by Chinese scientists,stands as a testament to the advancement of soybean agronomy and adaptability[8,9].Derived from parent cultivars originating in the Yellow River Basin, ZH13 boasts not only heightened genetic diversity but also a distinct ecological origin compared to the widely recognized Williams 82 cultivar.Herein, we generate ZH13-T2T, the T2T genome assembly of Chinese soybeans.With its unprecedented completeness and high quality, the assembly provides a superior reference genome, as well as a new opportunity to comprehensively decode and deeply understand the complex repeats in soybean genomes, which is invaluable to the society for cutting-edge plant genomics studies.Moreover, as the most planted soybean cultivar in China, the ZH13-T2T genome is also a valuable resource to molecular inbreeding.
Although efforts have been made, it is still a non-trivial task to implement high quality T2T genome assembly, as the employed assembly tools could still have bias and lead to mis-assemblies,while the read length could be limited to solve those extremely long repeats.During the generation of ZH13-T2T, we used several tailored approaches guarantee assembly quality.
One is the use of multiple assemblers to take their advantages and reduce bias.More precisely,the three sets of contigs independently produced by the Hifiasm,NextDenovo and Canu played different roles.Overall, the Hifiasm contigs reconcile accuracy and continuity,which were used as primary contigs.Some of NextDenovo contigs have even higher ability to span long repetitive regions so that they were employed to fill the unsolved regions.The Canu contigs are usually shorter due to the limited length of Hifi reads,however,they are accurate and useful to reveal the elements of difficult repetitive regions such as retrotransposon-or rDNA-rich loci.Moreover, Canu also has good performance in telomere regions.Thus, they also played an important role to guide gap filling, LCR correction and telomere refinement.
Another one is the monitoring of read coverage,which is effective to prevent mis-assembly.Theoretically, a perfect assembly should have uniform read coverage along the whole genome,especially with the low GC-bias of long read sequencing.Thus, abnormal read coverage is a good indicator to mis-assembly.During ZH13-T2T assembly,we used local read coverages to conduct quality control all the way, which not only helps to detect misassemblies, but also guide to correctly reconstruct the sequences of gaps, LCRs and HCRs.
Long repeats are still difficult to solve in practice, even if ONT ultralong data is available.We used an in-house tool to carefully collect and align anchored reads to iterative infer those extremely long repetitive sequences, with the guidance of the inherent sequence divergences and read coverages in local regions.The tool is able to improve the assembly in long repetitive regions, especially with known elements.However, it is still an open problem to develop more effective and generic tools to solve long repeats with limited read length.
The meticulous analysis of the previous gap regions within the soybean genome holds significant implications for soybean breeding.It is well established that soybean is a quintessential short-day plant and, as such, inherently exhibits sensitivity to photothermal conditions,particularly with regard to photoperiod.The responses to these photothermal conditions play a pivotal role in determining the soybean’s capacity for growth, development, yield formation,and its ability to thrive across varying geographical latitudes[4,67].Among these responses,flowering time and maturity stand out as the most influential factors dictating the geographical adaptability of soybean.A large number of quantitative trait loci and significant associated loci have been reported in the past, but they usually result in large candidate regions that make it hard to mine the causal genes, and there is a risk of missing candidate genes in a single reference genome.Our T2T-ZH13 reference genome annotation has unveiled a multitude of novel genes, offering promising prospects for the cloning of causal genes by providing fresh molecular targets.
Additionally, within these previously unexplored gap regions,we have annotated 505 novel genes.Through KEGG and GO enrichment analysis,it has come to light that these genes are implicated in a diverse array of biological pathways, encompassing various aspects of biosynthesis, metabolism, and cellular signal transduction.Simultaneously, they play pivotal roles in several biological functions, including gene regulation and RNA processing.These newfound genes hold the promise of serving as novel molecular targets for subsequent Genome-wide association studies (GWAS)and for the validation of related gene functions.
Beyond assembly improvements, ZH13-T2T enhances the soybean genome’s annotation, particularly regarding repetitive elements, centromeres, and telomeres.Repetitive elements play a significant role in genome evolution and gene regulation [68–71].We found that repeat elements constituted a significant portion of the genome, with retrotransposons, particularly LTR elements,being predominant.We also identified the presence of abundant satellites and rDNAs.The T2T genome exhibits a notable expansion in repetitive sequences compared to previous assemblies.Moreover, the identification and characterization of centromeres and telomeres within ZH13-T2T offer valuable insights into the organization and maintenance of chromosomal integrity.Soybean,a relic of ancient tetraploid plant evolution, has undergone two significant whole-genome duplication or polyploidization events [72].Within soybean genome, two distinct centromeric repeat classes exist,and their distribution is notably uneven,signifying the presence of two subgenomes in soybean.It is plausible that these subgenomes may have originated from the hybridization of two nowextinct plants with 2n = 20 chromosomes, followed by a subsequent partial homogenization of one centromeric repeat class by the other.Research findings suggest that the CentGm-1 ancestor possessed a higher chromosome count compared to the CentGm-2 ancestor[54].A thorough understanding of centromere structure is essential for breeding programs to develop soybean varieties with stable and accurate chromosome segregation.This ensures that the progeny of these plants have the correct number of chromosomes, preventing genetic abnormalities.
In conclusion, the ZH13-T2T genome represents a significant advancement in soybean genomics.The comprehensive genome annotation, identification of key genomic features, and insights into structural variations contribute to our understanding of soybean genetics and evolution.This high-quality reference genome will serve as a valuable resource for future studies in biology and practices in molecular breeding of soybean.
Code availability
Minimap2: https://github.com/lh3/minimap2;
SyRI: https://github.com/schneebergerlab/syri;
Barrnap: https://github.com/tseemann/barrnap;
INFERNAL: https://eddylab.org/infernal/;
MAFFT: https://mafft.cbrc.jp/alignment/software/;
EDTA: https://github.com/oushujun/EDTA;
RepeatModeler: https://www.repeatmasker.org/RepeatModeler/;
BUSCO: https://github.com/metashot/busco;
Augustus: https://github.com/Gaius-Augustus/Augustus;
TRF: https://github.com/Benson-Genomics-Lab/TRF;
TRF2GFF: https://github.com/Adamtaranto/TRF2GFF.
Data availability
The genome assembly data generated in this study can be achieved from NCBI with BioProject ID: PRJNA1015379 and BioSample accession: SAMN37355196.
CRediT authorship contribution statement
Anqi Zhang:Conceptualization, Formal analysis, Methodology,Visualization,Writing– original draft.Tangchao Kong:Conceptualization, Formal analysis, Methodology, Visualization, Writing –original draft.Baiquan Sun:Conceptualization, Formal analysis,Investigation, Methodology, Validation, Writing – original draft.Shizheng Qiu:Conceptualization, Formal analysis, Methodology,Visualization, Writing – original draft.Jiahe Guo:Conceptualization, Formal analysis, Methodology, Visualization, Writing – original draft.Shuyong Ruan:Conceptualization, Formal analysis,Methodology,Visualization,Writing–original draft.Yu Guo:Conceptualization, Formal analysis, Methodology, Visualization, Writing – original draft.Jirui Guo:Conceptualization, Formal analysis,Methodology, Visualization, Writing – original draft.Zhishuai Zhang:Conceptualization, Formal analysis, Methodology, Visualization, Writing – original draft.Yue Liu:Conceptualization, Formal analysis, Methodology, Visualization, Writing – original draft.Zheng Hu:Conceptualization, Formal analysis, Investigation,Methodology, Validation, Writing – original draft.Tao Jiang:Conceptualization, Formal analysis, Investigation, Methodology, Validation, Writing – original draft.Yadong Liu:Conceptualization,Formal analysis, Investigation, Methodology, Validation, Writing– original draft.Shuqi Cao:Conceptualization, Formal analysis,Investigation, Methodology, Validation, Writing – original draft.Shi Sun:Conceptualization, Formal analysis, Investigation,Methodology, Validation, Writing – original draft.Tingting Wu:Conceptualization, Formal analysis, Investigation, Methodology,Validation,Writing–original draft.Huilong Hong:Conceptualization,Formal analysis,Investigation,Methodology,Validation,Writing – original draft.Bingjun Jiang:Conceptualization, Formal analysis,Investigation,Methodology,Validation,Writing–original draft.Maoxiang Yang:Conceptualization,Formal analysis,Investigation,Methodology, Validation,Writing – original draft.Xiangyu Yao:Conceptualization, Formal analysis, Investigation, Methodology, Validation, Writing – original draft.Yang Hu:Conceptualization, Funding acquisition, Supervision, Writing – review &editing.Bo Liu:Conceptualization, Funding acquisition, Supervision, Writing – review & editing.Tianfu Han:Conceptualization,Funding acquisition, Supervision, Writing – review & editing.Yadong Wang:Conceptualization, Funding acquisition, Supervision, Writing – review & editing.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work has been supported by the National Key Research and Development Program of China (2021YFF1200105) and National Natural Science Foundation of China (62172125, 62371161).
Appendix A.Supplementary data
Supplementary data for this article can be found online at https://doi.org/10.1016/j.cj.2023.10.003.

- The Crop Journal的其它文章
- Flowering-time regulation by the circadian clock: From Arabidopsis to crops
- Global characterization of OsPIP aquaporins reveals that the H2O2 transporter OsPIP2;6 increases resistance to rice blast
- Drought-triggered repression of miR166 promotes drought tolerance in soybean
- The OsBSK1-2-MAPK module regulates blast resistance in rice
- Natural variation of an autophagy-family gene among rice subspecies affects grain size and weight
- Rice gene OsUGT75A regulates seedling emergence under deep-sowing conditions