
巨噬细胞极化在草酸钙肾结石形成中作用及其影响因素的研究进展*
2024-03-05 02:45:16李卫胜张传国李杨东何文强
中国病理生理杂志 2024年1期
李卫胜, 张传国, 李杨东, 何文强
(1河南中医药大学,河南 郑州 450003;2河南中医药大学第一附属医院泌尿外科,河南 郑州 450003;3新乡医学院第一附属医院泌尿外科,河南 卫辉 453100)
肾结石是泌尿外科多发性疾病,可造成肾功不全、肾萎缩和脓胸等并发症,给患者带来严重的经济负担[1-2]。草酸钙(calcium oxalate, CaOx)肾结石的形成涉及活性氧(reactive oxygen species, ROS)产生增多、免疫炎症反应和肾小管上皮损伤等病理过程[3]。既往研究证实,巨噬细胞的不同极化状态能够影响CaOx 肾结石形成[4]。M1 型巨噬细胞的过度激活会加剧肾脏组织的炎性损伤,与此有关的炎症因子可促进CaOx 晶体的生成,如干扰素γ(interferon-γ,IFN-γ)、肿瘤坏死因子α(tumor necrosis factor-α,TNF-α)、白细胞介素1β(interleukin-1β, IL-1β)等。与M1 型巨噬细胞不同的是,M2 型巨噬细胞主要通过吞噬晶体、分泌多种蛋白质和细胞因子如IL-4、IL-13、IL-10、转化生长因子β(transforming growth factorβ, TGF-β)等,来减轻炎症应激对肾小管上皮的损伤,促进肾脏组织的修复或再生,从而阻止CaOx 肾结石的形成。对调控巨噬细胞极化防治CaOx 肾结石的深入研究显示,巨噬细胞极化相关基因CXCL-14,以及人第11 号染色体上与消除ROS 有关的沉默信息调控因子3(silent information regulator 3,Sirt3)基因的过表达,能够诱导巨噬细胞发生M2 型极化,继而抑制CaOx 晶体的沉积[5-6]。本文从MPP 的调控机制及其功能、巨噬细胞极化与CaOx 肾结石发生机制的联系、特异性调控M1 或M2 型巨噬细胞极化防治CaOx 肾结石的机制进行综述,为CaOx 肾结石的特异性防治研究提供参考。
1 巨噬细胞极化的概述
巨噬细胞主要参与炎症调节、吞噬并杀伤入侵的病原微生物以及清除其他外源性异物等。在以上过程中,巨噬细胞的极化状态起到关键作用。巨噬细胞极化是巨噬细胞被体内外不同的微环境因子刺激,分化为表型及功能不一的M1 或M2 型巨噬细胞的现象,能够影响疾病的发生、发展和转归,如减轻炎症损伤、抑制肿瘤微环境等[7-8]。
M1 型巨噬细胞通过释放ROS、一氧化氮(nitric oxide, NO)和溶酶体酶参与病原体的杀伤和清除,分泌单核细胞趋化蛋白1(monocyte chemotactic protein-1, MCP-1)、IL-8、IL-6、TNF-α 等促炎细胞因子来引发炎症反应。不同的是,M2 型巨噬细胞通过释放TGF-β、IL-10等抗炎细胞因子减轻炎症应激,分泌血小板源性生长因子(platelet-derived growth factor,PDGF)、血管内皮生长因子(vascular endothlial growth factor, VEGF)、成纤维细胞生长因子(fibroblast growth factor, FGF)等促进血管形成、加速组织修复和创伤部位愈合[8-10],详细分类和功能见图1。
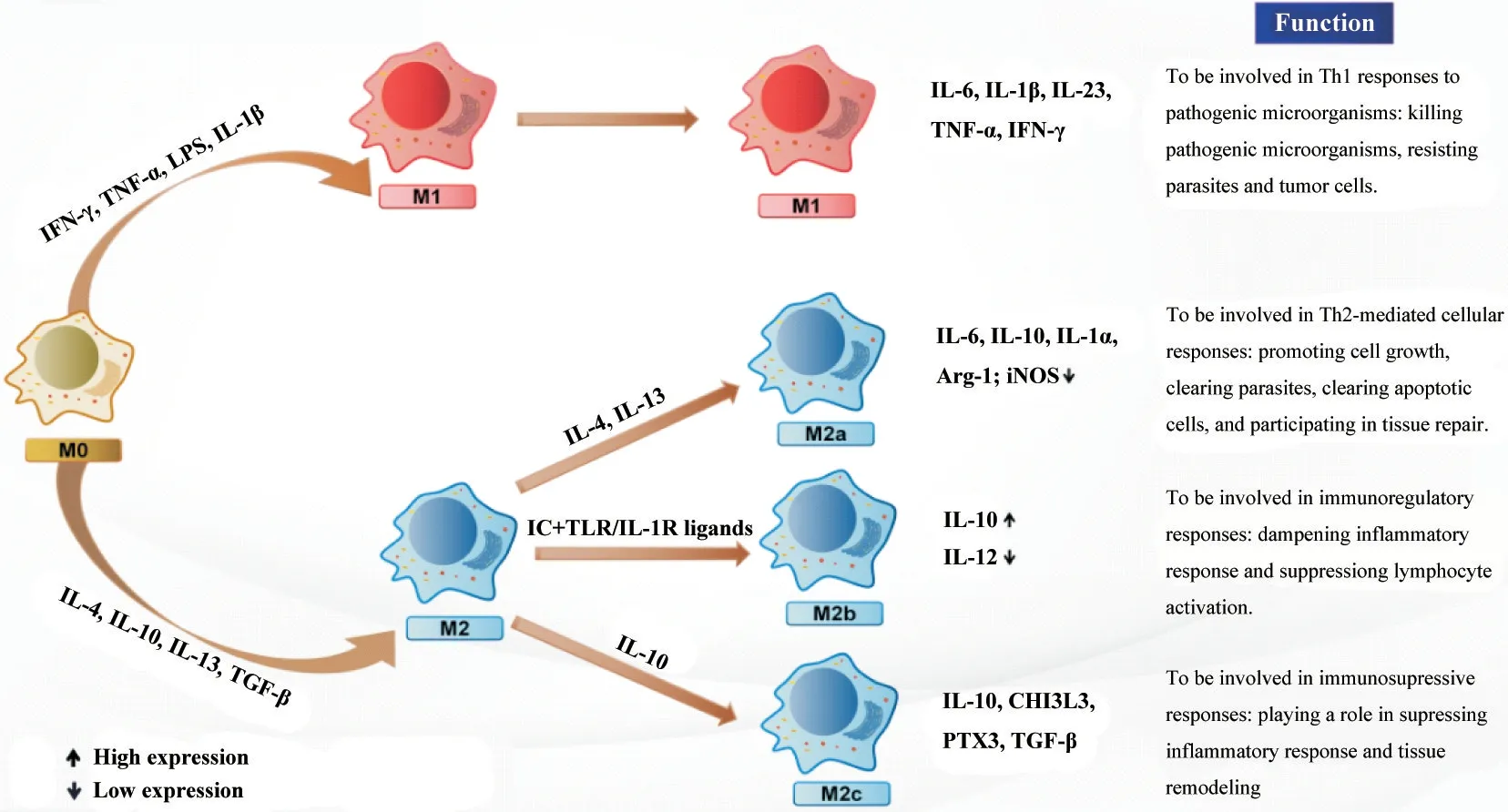
Figure 1. Regulation, classification and function of macrophage polarization. IFN: interferon; TNF: tumor necrosis factor; LPS: lipopolysaccharide; IL: interleukin; TGF: transforming growth factor; IC: immune complex; TLR: Toll-like receptor; IL-1R: interleukin-1 receptor; Arg-1: arginase-1; iNOS: inducible nitric oxide synthase; CHI3L3: chitinase 3-like protein 3; PTX3: pentraxin 3.图1 巨噬细胞极化的调控、分类和功能
1.1 M1 型巨噬细胞极化的调控机制 M1 型巨噬细胞主要受脂多糖(lipopolysaccharide, LPS)、IL-1β、IL-6、TNF-α 和IFN-γ 等细胞因子的调控。这些细胞因子通过结合不同受体,如Toll 样受体(Toll-like receptors, TLRs),激活细胞内下游信号通路,从而促进M1 型极化、分泌和增殖。TLRs 是较为重要的细胞因子受体,且TLR2和TLR4与M1型巨噬细胞的关系最为密切。LPS 和病原相关分子模式(pathogenassociated molecular pattern, PAMP)分别与TLR2 和TLR4 相结合,都通过诱导干扰素β 的含TIR 结构域街接蛋白(TIR domain-containing adaptor inducing interferon β, TRIF)和髓样分化因子88(myeloid differentiation factor 88, MyD88)信号通路促使一系列核因子进入细胞核,如核因子κB(nuclear factor-κB, NFκB)、干扰素调节因子3(interferon regulatory factor 3,IRF3)、活化蛋白1(activator protein-1, AP-1)等,从而调控DNA 的转录,增加M1 型巨噬细胞分泌IL-1β、TNF-α、IFN-γ等炎症因子[10-11],详见图2。
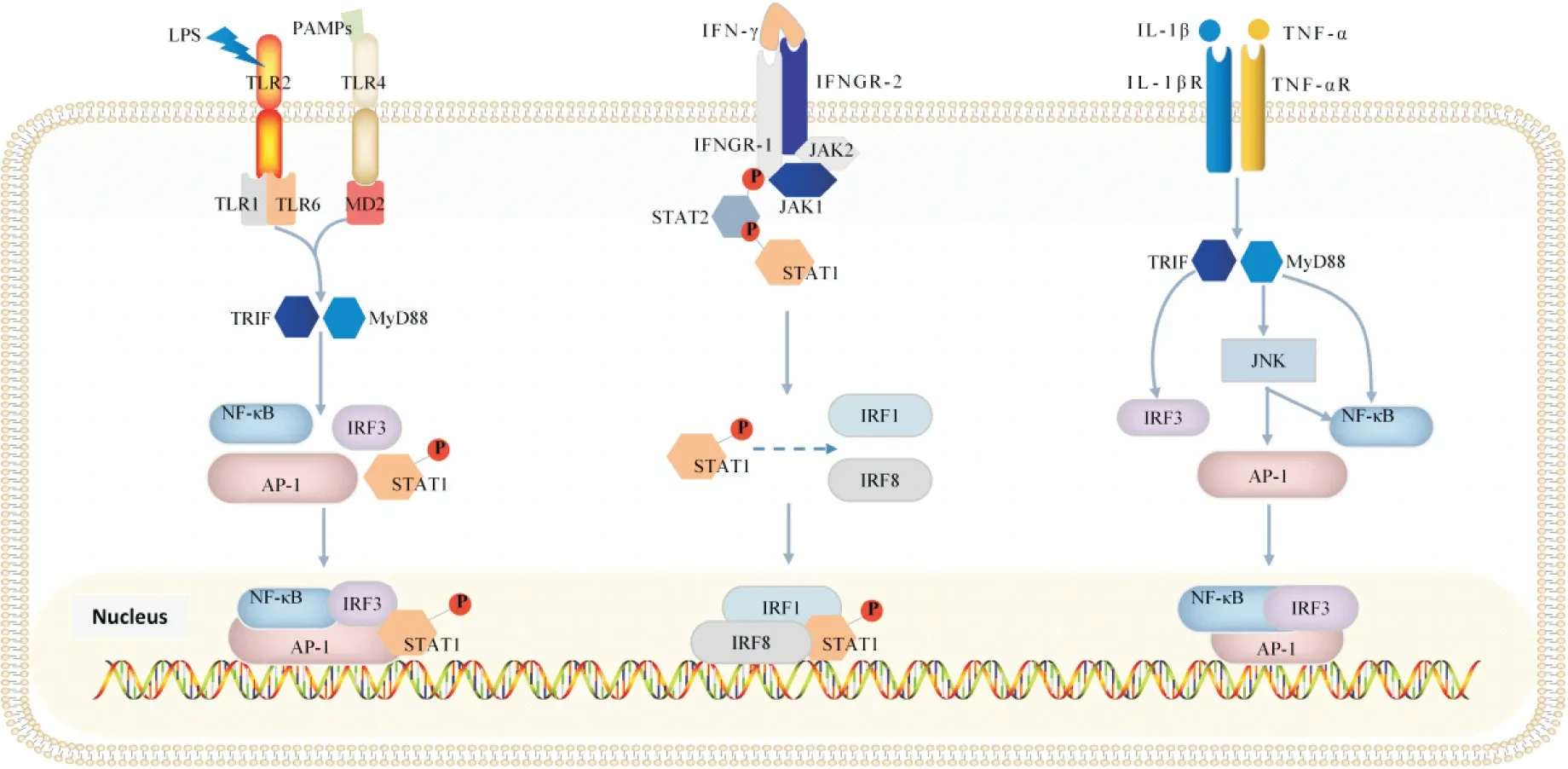
Figure 2. Regulatory mechanisms of M1 macrophage polarization. Toll-like receptor 1(TLR1) and TLR6, which act as chaperone proteins for TLR2, emerge as key participants in this intricate process of the regulatory mechanisms that govern the polarization of M1 macrophages. In addition, TLR4 is a chaperone protein for myeloid differentiation protein 2(MD2). TIR domain-containing adaptor-inducing interferon (TRIF) and myeloid differentiation factor 88 (MyD88) signaling pathways are initiated by the coordinated activation of TLR2 and TLR4, which significantly affects DNA transcription through nuclear factors. The biological activities of M1 macrophages and their polarization are ultimately regulated by these complex regulatory networks. Notably, the interplay between the transcription factors interferon regulatory factor 1 (IRF1) and signal transducer and activator of transcription 1 (STAT1) plays a crucial part in starting DNA transcription.图2 M1型巨噬细胞极化的调控机制
1.2 M2 型巨噬细胞极化的调控机制 M2 通常被一些细胞因子或免疫复合物激活,如巨噬细胞集落刺激因子(macrophage colony-stimulating factor, MCSF)、IL-4、IL-10、IL-13 和TGF-β 等,主要参与调控免疫应答、组织修复等。IL-4 和IL-13 是M2 型极化的主要调控因子,通过激活JAK-STAT6 途径促进M2型极化[13]。此外,IL-4 还通过激活PI3K-AKT-mTOR途径促进M2 型极化。研究表明,mTOR 有两种复合体,即mTORC1 和mTORC2,其中mTORC1 具有调节代谢、抑制降解、储存能量和维持细胞生长的功能,IL-4 诱导增殖和调节代谢的作用,主要与mTORC1有关[14-15]。IL-10 作为体内一种重要的免疫抑制因子,主要由M2 型巨噬细胞分泌,通过激活JAKSTAT3途径促进M2型极化[16],详见图3。
2 巨噬细胞极化与CaOx 肾结石形成机制的潜在联系
近年来,巨噬细胞极化在CaOx 肾结石形成中的作用被广泛关注,其研究主要集中在巨噬细胞的表型转化[17-18]。CaOx 晶体可引起炎症应激的发生,并抑制M2型巨噬细胞吞噬晶体的能力,从而导致肾小管上皮细胞受损,使Randall 斑块暴露到尿液中成为结石形成的基础[19]。Taguchi 等[20]的研究显示,Randall 斑块及其周围组织中促炎因子的基因表达显著上调,同时伴有M1 型极化的增加和M2 型极化的减少[4]。将CaOx 晶体与肾小管上皮细胞共培养后,肾小管上皮细胞显著表达MCP-1、骨桥蛋白(osteopontin, OPN)和TNF-α 等炎症标志物[21-22]。在上述实验基础上加入巨噬细胞和脂肪细胞,并观察CaOx 晶体对肾小管上皮细胞的黏附情况。结果显示,CaOx 晶体对肾小管上皮细胞的黏附性增加,提示三种细胞之间可能存在某种旁分泌机制,这种机制可诱导肾小管发生炎性损伤,MCP-1、OPN 和TNF-α 的mRNA表达升高是关键因素。值得关注的是,MCP-1、OPN和TNF-α 等炎症标志物的mRNA 表达水平可能是实现个体化精准治疗的关键,特异性调控巨噬细胞极化为肾结石疾病的防治开辟了新的研究方向。
3 基于巨噬细胞功能防治CaOx 肾结石的机制探索
正常生理状态下,机体在促炎与抗炎之间保持着微妙的平衡状态。例如,肾小管上皮细胞具有吞噬CaOx 晶体的能力,后者又可以刺激肾小管上皮细胞表达高迁移率族盒蛋白1(high mobility group box protein 1, HMGB1)、MCP-1 等炎症相关蛋白,这实际上启动了巨噬细胞的分化过程。尿液中CaOx 晶体的过饱和状态能够引发ROS 超载和炎症应激,导致肾脏组织损伤;相反,巨噬细胞可以通过调节免疫炎症反应来减轻肾脏损害[23-24]。然而,肾内发生炎症应激会破坏肾小管的结构和功能,导致其排泄和分泌功能障碍,造成CaOx 晶体长期滞留于肾小管,增加了患肾结石疾病的风险。Taguchi 等[25]的研究显示,M2 型巨噬细胞与M1 型巨噬细胞相比具有更强的吞噬和降解晶体的能力,可以有效阻止CaOx 晶体的形成。巨噬细胞极化在免疫调节中的生物学作用,为CaOx 肾结石的防治提供了有价值的思路。关注M2型巨噬细胞极化的调控研究似乎更易于推动肾结石治疗的发展。通过诱导巨噬细胞向M2 型极化来减轻炎症应激对肾脏组织的损伤,是未来防治CaOx 肾结石的重要策略。
3.1 M1 型巨噬细胞极化防治CaOx 肾结石的机制探索 Yu 等[26]对CaOx 肾结石患者的血样进行了综合分析。结果显示,M1 相关促炎性趋化因子(如TNF-α、IL-1 和IL-1β)的表达显著上调,且M1/M2 比值升高,提示M1型巨噬细胞在促进氧化应激诱导的肾脏损伤中起关键作用,增加了CaOx 肾结石的患病风险。这种现象可能与巨噬细胞的吞噬活性有关。巨噬细胞吞噬作用发生后,在NADPH 氧化酶或NAD氧化酶的作用下会产生ROS,用于消灭病原菌,但ROS 产生过剩会增加核苷酸结合寡聚化结构域样受体蛋白3(nucleotide-binding oligomerization domainlike receptor protein 3, NLRP3)炎症小体受体的表达,从而通过启动NF-κB 炎症通路,释放大量的炎症因子。这些事件最终导致肾脏损害,并促进了Randall 斑块的形成,以及CaOx 晶体的沉积和聚集[27-30]。CaOx 晶体与烯醇化酶1之间的相互作用可促进单核细胞迁移,通过抑制NAD+依赖性脱乙酰酶Sirt3的表达或激活NLRP3炎症小体,引起促炎介质的增加,从而促进肾脏内的炎症反应和纤维化过程[9]。
在使用罗格列酮(rosiglitazone, ROSI)治疗后,肾小管损伤、晶体黏附性及氧化应激反应等明显减轻,同时M1 型极化明显减少,M2 型极化显著增加;ROSI 调控巨噬细胞极化、抑制氧化应激及减轻炎性损伤的作用通过Nrf2/HO-1 信号通路介导[31-32]。因此,调节巨噬细胞极化的药物制剂对于CaOx 肾结石的防治具有重要意义。此外,应用ROS 抑制剂或抗氧化剂可有效阻止M1型巨噬细胞诱导的炎性损伤,是防治CaOx肾结石的可行途径。
3.2 M2 型巨噬细胞极化防治CaOx 肾结石的机制探索 Xi 等[11]的研究显示,Sirt1 通过将FOXO1 去乙酰化诱导M2 型极化。M2 型巨噬细胞则通过下调NADPH 氧化酶的激活水平,减轻肾小管上皮细胞的氧化应激损伤。此外,M2 型巨噬细胞也能够直接抑制NADPH 氧化酶的激活,从而减少ROS 的产生和p38 MARK 的表达,并通过增加Akt 的磷酸化,增强细胞的增殖和修复能力,进一步减轻组织损伤[18]。上述研究为调控M2 型极化来抑制CaOx 晶体沉积的研究提供了依据。
M-CSF 可以激活M2 型巨噬细胞,从而增强其吞噬CaOx 晶体的能力。然而,肾脏中雄激素受体(androgen receptor, AR)-miR-185-5p/CSF-1 途径的激活抑制了CSF-1 的表达,导致M2 型巨噬细胞的活化状态及其吞噬能力下降,易于结石形成[33-34]。M2 型巨噬细胞极化在减轻炎症应激造成的肾小管上皮损伤及促进组织修复方面发挥了重要作用,并通过吞噬晶体、促进结晶排泄等减少CaOx结晶的沉积。
3.2.1 Sirt1 诱导M2 型巨噬细胞极化预防CaOx 肾结石的作用 草酸盐可导致ROS 的生成增加,从而影响单核细胞的线粒体功能,促使mRNA 的不稳定性和细胞凋亡数增加,并诱导巨噬细胞向M1 型极化。Sirt1是sirtuin家族中已被广泛研究的一种NAD+依赖性脱乙酰酶。近来研究表明,Sirt1 的表达在炎症相关疾病中受到抑制,而Sirt1 的过度表达增强了细胞的抗氧化应激和抗炎特性[35]。在CaOx 肾结石的发生发展过程中,Sirt1主要通过诱导M2型巨噬细胞极化,参与巨噬细胞分化的动态变化。Song 等[36]研究显示,过表达Sirt1 的巨噬细胞倾向于M2 型极化,能够显著抑制高草酸尿小鼠肾脏细胞的凋亡,减轻肾脏损伤。然而,经草酸钙预处理的巨噬细胞中Sirt1 的表达明显减少,从而激活Notch 信号通路,促进巨噬细胞向促炎M1 表型极化。过表达Sirt1 或抑制Notch 信号通路的激活可能是预防肾结石疾病的潜在靶点。
3.2.2 过氧化物酶体增殖物激活受体γ(peroxisome proliferator activated receptor-γ, PPAR-γ)诱导M2 型巨噬细胞极化预防CaOx 肾结石的作用 柯里拉京(corilagin)是一种半乳糖苷,具有抑制ROS 生成和减轻线粒体DNA 氧化损伤的作用,还具有强大的抗炎作用。在Yuan 等[37]的研究中,给予CaOx 肾结石大鼠模型使用不同剂量的corilagin后,氧化应激标志物丙二醛(malondialdehyde, MDA)的水平显著降低,抗氧化酶的分泌增加,并随corilagin 浓度的升高呈梯度式增加。通过实时荧光定量PCR 检测细胞存活标志物的mRNA 水平,显示结石组PPAR-γ、PI3K和Akt 的mRNA 表达显著降低,而corilagin 组三者的表达水平显著升高,提示corilagin 可能通过PPAR-γ和PI3K/Akt 介导的通路抑制CaOx 晶体诱导的氧化应激和炎症反应。值得注意的是,PPAR-γ 激动剂(如吡格列酮和ROSI)在治疗肾结石方面显示出有效性。这些激动剂通过促使M2型巨噬细胞极化,有效减少肾脏中CaOx 晶体的沉积,减轻炎症损伤[34]。研究表明,PPAR-γ 通过上调miR-23,从而减弱干扰素调节因子1(interferon regulatory factor 1, IRF1)和Pknox1 的表达,使M2 型极化增强[38]。此外,PPAR-γ的激活导致CD206+巨噬细胞数量增加,继而通过分泌IL-10 促进M2 型极化[39]。因此,PPAR-γ 的激活在抑制CaOx肾结石的形成中起着关键作用。
3.2.3 核因子E2相关因子2(nuclear factor E2-related factor 2, Nrf2)/血红素加氧酶1(heme oxygenase-1,HO-1)诱导M2 型巨噬细胞极化预防CaOx 肾结石的作用机制 Nrf2 是人体抗氧化应激损伤的主要的转录调节因子,在急性肾损伤、糖尿病肾病以及狼疮性肾炎等多种肾脏病中发挥重要的保护作用。最近的研究显示,慢性肾脏病进程中肾脏纤维化指标越高,Nrf2 和谷胱甘肽过氧化物酶4(glutathione peroxidase 4, GPX4)的表达越低,提示Nrf2/GPX4 信号通路被抑制[40]。在使用Nrf2 激动剂后,慢性肾脏病患者的肾纤维化得到缓解,铁死亡被抑制,说明激活Nrf2/GPX4信号通路具有保护肾脏的作用,而Nrf2是关键调节因子[40]。Wang 等[41]证实,昼夜节律基因BMAL1在维持机体正常稳态中不可或缺,过表达BMAL1基因能够激活核Nrf2/HO-1 信号通路,从而减轻草酸盐诱导的氧化应激损伤,减少尿CaOx 结石的形成。ROSI 由于其显著的抗氧化应激特性和调节巨噬细胞极化的能力,成为治疗和预防CaOx 肾结石的潜在治疗剂。体外实验显示,ROSI 显著上调M2 型巨噬细胞标志物精氨酸酶1(arginase-1, Arg-1)和CD206,以及抗炎细胞因子IL-4 和IL-10 的表达[32]。ROSI 主要通过激活HK-2 细胞中的Nrf2/HO-1 途径抑制氧化应激,从而减少肾小管的炎症性损伤[32]。Nrf2 激动剂及其信号通路的激活为尿路结石的防治开辟了新方向。
4 总结与展望
本文综述了肾脏中巨噬细胞与肾结石形成的复杂关系,以及巨噬细胞极化在这些结石发生机制中的关键作用,并涉及巨噬细胞极化与CaOx 肾结石发生有关的基因遗传学研究。值得注意的是,M2 型巨噬细胞在肾结石发生时比M1 型巨噬细胞的吞噬功能更好,能够加快尿液中草酸盐晶体的分解和代谢,从而减少CaOx 晶体的沉积,特异性增强M2 型极化是更具可能性的治疗靶点。
此外,肾结石发生的个体差异与基因遗传易感性有关,很多基因或分子途径已被确定为患肾结石疾病的潜在危险因素。巨噬细胞在不同个体或同一个体不同器官的组织中具有异质性,这也与基因有关。M1 型巨噬细胞相关基因的表达增加了肾结石疾病的患病风险,而M2型巨噬细胞相关基因的表达与抑制结石的形成有关。揭示单核吞噬细胞群、基因遗传易感性和CaOx 肾结石之间的复杂关系在绘制新的研究轨迹方面有巨大的前景。然而,目前的研究未能充分阐明单核吞噬细胞群、基因遗传易感性和CaOx 肾结石之间复杂的相互作用。在未来研究中进一步探究此问题,有助于发现CaOx 肾结石的潜在治疗靶点。
猜你喜欢
现代财经-天津财经大学学报(2022年5期)2022-06-01 06:08:32
中成药(2021年5期)2021-07-21 08:39:02
基层中医药(2020年6期)2020-09-11 06:35:26
现代临床医学(2019年6期)2019-12-07 06:03:50
电子测试(2017年15期)2017-12-18 07:18:51
中外医疗(2015年11期)2016-01-04 03:58:45
电源技术(2015年1期)2015-08-22 11:16:18
医学研究杂志(2015年8期)2015-06-22 14:00:57
医学研究杂志(2015年12期)2015-06-10 06:57:46
西南军医(2015年6期)2015-01-23 01:25:49
