
HIV 非核苷酸逆转录酶抑制剂的合成
2024-02-29 09:35:08王奕程
山西化工 2024年1期
王奕程
(浙江省义乌中学,浙江 金华 322000)
0 引言
获得性免疫缺陷综合症(AIDS)是当今世界最为棘手的传染病之一,它由人类免疫缺陷病毒(HIV)引起。获得性免疫缺陷综合症拥有漫长的潜伏期(平均8~9 年),这对疾病的诊断增加了困难。除此之外,目前尚未有一种高效的药物治疗获得性免疫缺陷综合症,而是采用通过多种药物的联合作用来达到治疗目的。病毒耐药性对药物开发带来巨大的挑战,不过随着对病毒感染机制以及药物的研究的深入,获得性免疫缺陷综合症已经从致命传染病转变可控的慢性传染病。
逆转录酶是HIV 乃至几乎所有RNA 病毒最微妙且重要的结构,它是病毒增殖的核心。因此许多抗HIV 的药物是针对逆转录酶的,这被称为高活性抗逆转录病毒疗法(HAART)[1]。其中,非核苷酸逆转录酶抑制剂,是HAART 的一类重要药物。它们通过与HIV-1 逆转录酶p66/p51 异二聚体p66 亚基上的单个位点结合而与HIV-1 逆转录病毒相互作用[2]。截止2020 年,共有6 种FDA 批准的非核苷酸逆转录酶抑制剂[1]。非核苷酸逆转录酶抑制剂的结构和功能是高度统一的,它们有多个芳香环,两侧结构类似于鸟的翅膀。因此它们拥有构象灵活性和位置适应性,以对突变形成高度的遗传屏障[3]。
本文针对现有的三种非核苷酸逆转录酶抑制剂类药物Rilpivirine、Etravirine 以及Doravirine 的合成路线进行研究,通过分析反应条件、反应机理以及最终产率,寻找更为高效的合成路线。最优化的过程不仅仅局限于对反应路线的改进,还有在反应路线的基础上对反应参数的最优化,目的是寻找总产率更高、操作更便捷、环境更友好的有潜在大规模应用价值的合成路线。这三种药物分子的结构如图1。
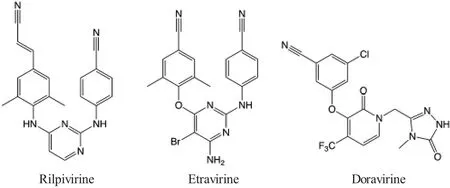
图1 三种重要的非核苷酸逆转录酶抑制剂
1 Rilpivirine 的合成
在上述三种非核苷酸逆转录酶抑制剂中,Rilpivirine(RPV,Cabenuva)是其中相对较为简单的分子。Ripivirine 是一种研究较为深入的非核苷酸逆转录酶抑制剂,不论是在分子动力学模拟上(PDB ID 4G1Q),还是在化学合成上。Rilpivirine 具有几乎所有非核苷酸逆转录酶抑制剂共有的链状芳香环结构,使药物拥有更高的柔性,可以更加灵活并高效地与逆转录酶上的活性位置特异性结合。传统的Rilpivirine 合成存在一些问题,不仅效率低下,所花费的时间长,而且原料成本相对昂贵,更存在一些污染环境的问题,通常传统合成的产率为18.5%[4]。而在以下的合成中,不仅产率提高为21%,而且在反应时间、原料成本以及绿色化学角度上,都有着显著的提升[4]。
Rilpivirine 是一个没有手性中心的分子,并不涉及立体选择性带来的不方便。通常可以通过逆合成分析,将它切断为两部分,即左侧部分4 与右侧部分8(如图2)。这一步切断是可靠的,这得益于嘧啶环的亲电性,因为氮原子的高电负性可以有效稳定负电荷,促进芳香杂环化合物的亲核取代反应。离去基团可以是卤素原子或者其他的离去基团,通常选择易于制备的氯离去基团。

图2 Rilpivirine 的合成路线[4]
合成左侧部分4 的核心在于构建苯环上α,β-不饱和腈基,这里有两种常见的思路。第一种是通过经典的苯环上的芳香亲电取代反应。例如先采用Friedel-Crafts 酰化反应,在还原脱水可以得到稳定的共轭体系(常用的还原剂可以是硼氢化钠、硼氢化锂,或者是更为温和的氰基硼氢化钠和硼烷);或者采用类似于Friedel-Crafts 酰化反应的Gattermann-Koch反应,利用酰胺钝化的氨基与一氧化碳-氯化氢混合物反应,酰胺水解后伴随羟醛缩合反应合成左侧片段。但是容易预见的是这两条路线并不理想,因为至少4 步以上的操作会导致产率的降低。因此第二种思路便是采用直接的有机金属催化反应,即Heck 反应。这也是许多Rilpivirine 合成所青睐的反应。传统的合成中,第一步通过酰胺与碘取代芳香环反应,在醋酸钯的催化下得到79.5%的产率[4]。这一步是不经济的,因为醋酸钯催化剂的循环使用效率较低,以及接下来还需要用脱水剂将酰胺脱水造成的产率偏低。改进后的合成虽然同样使用Heck 反应,却将反应物酰胺改成了α,β-不饱和腈2,将催化剂醋酸钯改成了钯-碳催化剂[4]。这条路线不仅将两步反应缩短为一步,而且钯-碳催化剂可以被循环利用,很大程度上解决了反应经济性的问题,中间体3 的产率为81%,如图2[4]。
合成右侧部分的核心在于构建嘧啶环,合成嘧啶环的思路是直接的,通过胍及其衍生物与1,3-二羰基化合物的反应。这一步合成嘧啶环的标准反应受热力学控制,并不需要对反应体系施加过多的影响便可以得到正确的产物,这得益于嘧啶酮环的芳香性。合成路线中采用1-(4-氰基苯基)胍5 和2-(乙氧基亚甲基)丙二酸二乙酯6 反应,先是合成芳香环的反应,在100 ℃的条件下受热力学控制得到7,伴随着酯的水解与脱羧反应[4]。最后,是嘧啶酮环与三氯氧磷的氯化反应,这是在嘧啶、吡啶等杂环中通用的,得到中间体8,如图2。
最后,左右两部分发生芳香亲核取代反应,左侧部分的氨基氮原子进攻右侧部分嘧啶环上带有氯原子离去基团的碳原子,生成最终产物Rilpivirine。这一步中利用的微波辐射法是Gedye 和Giguere 在1986年的研究中被提倡的,因为它能在一定程度上缩短化学过程所需的时间,提高反应的效率[5-6]。微波辐射下在乙腈中反应90 min,得到Rilpivirine 9,总产率为21%[4]。
2 Etravirine 的合成
Etravirine(ETV,Intelence)拥有较高的特异性、较强的活性以及较低的毒性,是典型的第二代HIV 非核苷酸逆转录酶抑制剂,并且总是与其他HIV 药物联合使用。然而,现有的Etravirine 的合成路线与Rilpivirine 一样存在一些缺点,例如反应效率低下,所花费的时间长等。因此,开发新的合成路线是十分有必要的。
Etravirine 在骨架上与Rilpivirine 十分相似,同样有着中间的嘧啶环与左右两侧含有苯环的侧链。因此,合成Etravirine 的方法应当与Rilpivirine 相似。对Etravirine 实施类似于Rilpivirine 的切断,得到1-(4-氰基苯基)胍、经过化学修饰的丙二酸二乙酯以及4-羟基-3,5-二甲基苄腈。4-羟基-3,5-二甲基苄腈的4 号位酚羟基在弱碱性条件下失去氢离子形成具有亲核性的氧负离子,进攻类似于图2 中间体8 中的氯原子得到目标产物Etravirine。这是一种高效的Etravirine 合成,它构成了下述的第二类Etravirine 合成。
化学上常用的Etravirine 合成有两类,第一类是从取代的嘧啶环出发的合成[7],第二类是从取代的胍出发自行构建嘧啶环的合成[8]。
在第一类合成中,从容易制备的2,4,6-三氯嘧啶10 出发,由于嘧啶环中含有高电负性的氮原子,提高了2,4,6-三氯嘧啶的亲电性,因此其作为亲电试剂。同时,在N,N-二异丙基乙胺(DIEA)的碱性环境下,4-羟基-3,5-二甲基苄腈中的酚羟基失去氢原子并作为亲核试剂,发生芳香亲核取代反应,生成中间体11。接下来嘧啶环再次作为亲电试剂与4-氨基苯甲腈发生第二次芳香亲核取代反应,生成中间体12。这一步是一个竞争反应,有两个合理的亲电位点,即亲核试剂可能进攻任意一个带有氯原子的碳原子,因此产物是两种亲核取代反应产物的混合物。幸运的是,在合适的实验控制下,通过降温(通常在-15~-10 ℃)并使用DMSO、DMF 等作为溶剂,在碱性或弱碱性条件下可以避免副产物的形成[7],并通过重结晶的方法分离得到中间体12。最后12 作为亲电试剂与氨气通过氨化得到13。至此嘧啶环上有三个给电子的取代基团,因此嘧啶环被活化,在没有Lewis 酸的条件下直接与溴水发生芳香亲电取代反应得到目标产物14,总产率为30.4%[7],合成路线如图3。
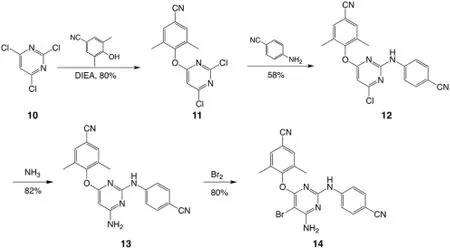
图3 从取代的嘧啶环出发的Etravirine 合成路线[7]
在第二类合成中,从4-胍苄腈15 出发,胍上2 个氮原子的孤对电子进攻丙二酸二乙酯的羰基碳原子,在乙醇钠与乙醇中通过反应顺序复杂的热力学控制的缩合反应生成具有芳香性的稳定的嘧啶环,用三氯氧磷氯化得到中间体17。同样,17 被活化基团取代,在没有Lewis 酸的条件下与溴水反应得到溴代产物18。再通过与第一类合成类似的芳香亲核取代反应生成主要产物19,氨化后得到目标产物,总产率为9.4%[8],显著低于第一类合成,合成路线如图4[8]。
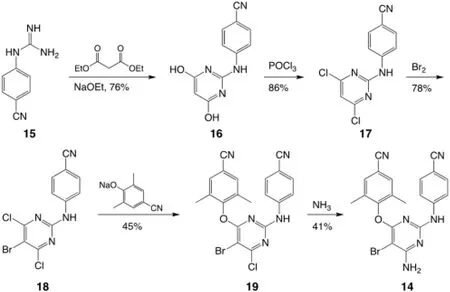
图4 从取代的胍出发的Etravirine 合成路线[8]
由于第一类合成对比与第二类合成产率的显著优势(30.4%与9.4%),因此许多合成最优化的研究都致力于提升第一类合成的产率。以下的合成条件(如图5),通过采用微波辐射促进的方法促进图三中间体12 到13 的转化,很大程度上提升了反应的效率(从原先的12 h 缩短至15 min。通过对反应参数的正交实验,确定了130 ℃与15 min 的最佳反应参数。最终,12 到13 的氨化反应的产率从原先的82.7%~83.6%提升至85.6%,是利用微波辐射促进方法的成功案例[8]。
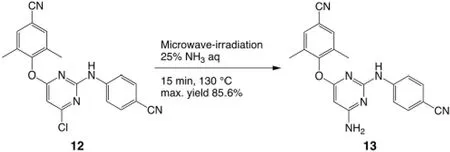
图5 微波辐射方法促进中间体3 的氨化反应[8]
3 Doravirine 的合成
在本文中所涉及的4 种非核苷酸逆转录酶抑制剂中,Doravirine(DOR,Pifeltro)是结构上较为复杂的一种。这主要是因为位于右侧的1,2,4-三唑环状结构。Doravirine 是一种具有良好耐受性并且在初期的高活性抗逆转录病毒疗法中发挥重要作用的药物。与Etravirine 类似,Doravirine 需与其他抗逆转录病毒药物联合使用,用于无既往抗逆转录病毒药物治疗史的成年HIV-1 感染患者。Doravirine 是一款在血脂上有临床获益的新型非核苷逆转录酶抑制剂,能够减缓HIV 感染者的体重增长并降低心血管疾病、糖尿病、高血压等疾病的风险。同时,Doravirine 十分特异,很少有脱靶效应。由于Doravirine 在高活性抗逆转录病毒疗法中的种种优势,如何高效地合成Doravirine 成为了必须解决的问题。最初Doravirine 的合成路线是为了给Doravirine 临床前毒性研究提供原料,但是这条路线存在多种缺陷,例如复杂的官能团操纵。而Merck 公司的以下的Doravirine 合成路线在多方面对传统的合成进行了改进,并在总产率上得到了很大的提升,为52%[9]。
Doravirine 中的取代的1,2,4-三唑是位于右侧的关键结构,它拥有芳香性,因此可以采用热力学控制的芳香杂环化合物合成的方法构建。首先从20 出发,经过简单的加成-消除反应生成酰胺21,并于肼的水合物在异丙醇(IPA)中生成22,22 不经过分离直接与羧酸发生第二个加成-消除反应生成酰胺23。此后是在热力学控制的条件下(90 ℃)的缩合反应,由23 中左侧羰基左侧的氮原子进攻右侧的羰基生成亚胺,即具有芳香性的1,2,4-三唑24。最后,在氯化亚砜的作用下羟基转化为氯原子,成为亲电试剂25,为最后的分子组装作准备,如图6[9]。合成Doravirine 中的取代的1,2,4-三唑的产率为57.5%[9]。在Merck 公司改进后的合成中,对1,2,4-三唑的合成是没有太大改动的。
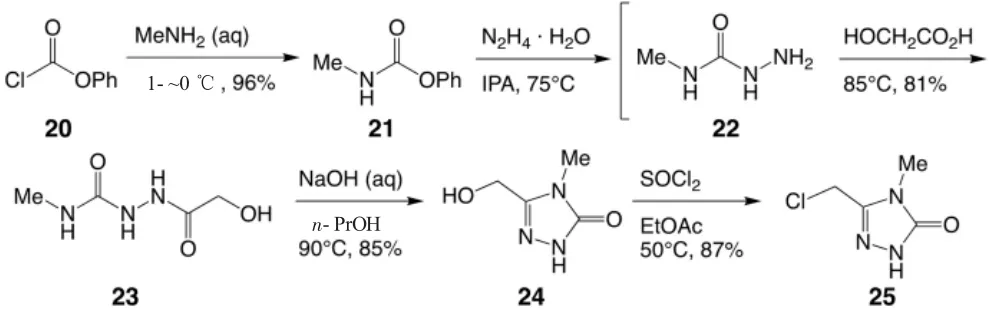
图6 Doravirine 中间体取代的1,2,4-三唑合成路线[9]
传统的合成是利用中间体25 合成最终产物Doravirine 30 的过程。第一步是经典的用金属锌生成烯醇盐衍生物的Reformatsky 反应,在这里选择烯醇盐的衍生物是为了防止副反应的发生,在四氢呋喃中生成缩合产物28。28 过氨化以及环化,生成吡啶酮29。29 先利用酰胺上的氮原子对被保护的1,2,4-三唑中间体25 的氯原子发生亲核取代,伴随着在弱碱性环境下酚羟基上的氧原子对吡啶酮上带有氟原子的碳原子的芳香亲核取代反应。这一步SNAr 反应在反应机理上是有利的,得益于缺电子的吡啶酮环以及高电负性的氟原子活化的碳原子,得到目标产物30,如图7[9]。
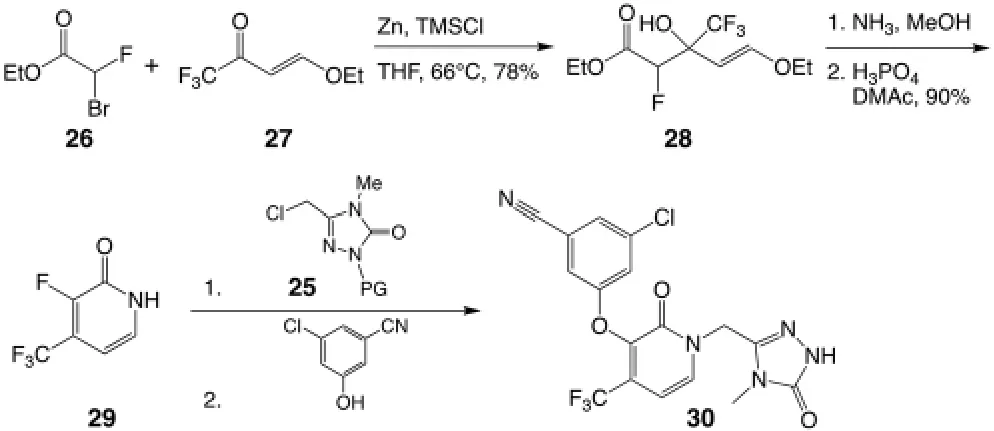
图7 Doravirine 的合成路线[9]
尽管图7 中的合成路线是十分有效的,但是为了进一步提高产率与高效性,不仅需要在反应顺序上进行调整,还要对离去基团(例如28 中的OEt 基团)、溶剂、温度、pH 值、浓度、化学计量数等反应参数进行最优化调整。对离去基团的研究让许多有机化学合成团队对OEt 离去基团进行研究,因为它有较大的提升空间与应用价值。Merck 公司的合成最优化研究提出了从中间体31 和32 出发,以甲苯作为溶剂,在1.7 mol/L的KOtAm(叔戊醇钾)和三乙胺中经过羟醛缩合反应合成中间体二烯33,在反应混合物中加入三氟乙酸酐,33 不经过分离直接氨化后环化,被转化为芳香杂环化合物吡啶酮34[9]。具体的反应参数如图8,从31出发到34 的这两步反应的总产率高达68%[9]。随后,图6 中合成的1,2,4-三唑中间体25 与新合成的34发生亲核取代反应,得到Doravirine 30,这一步产率为90%[9]。最终,Doravirine 30 不仅在化学纯度上有所提高,总产率也高达52%,这一合成可以作为一种可靠的Doravirine 合成方法[9]。
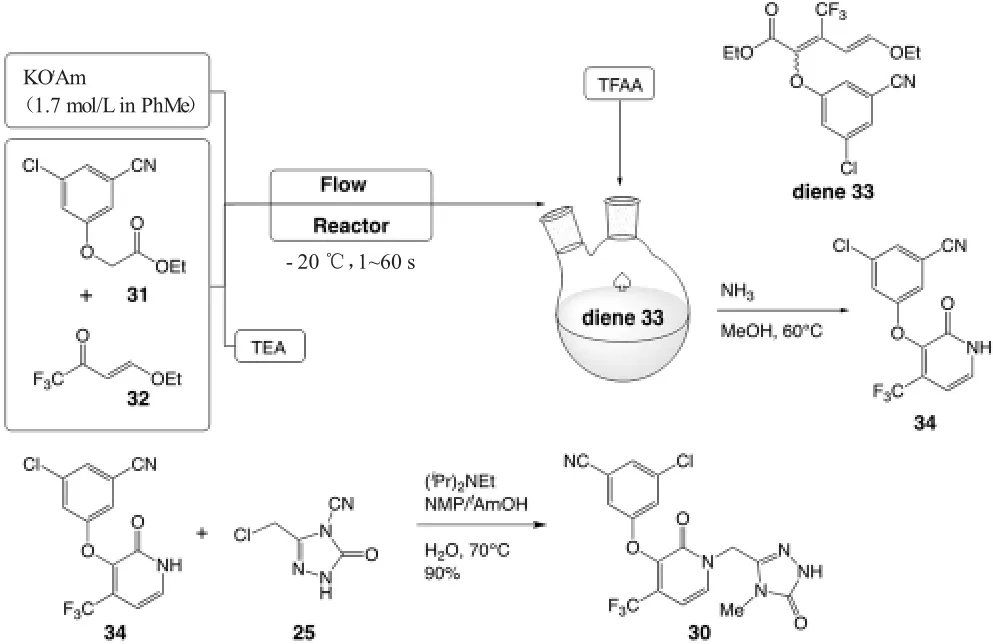
图8 Merck 公司对中间体34 合成所做的最优化改进[9]
4 结论
本文通过对3 个经典的HIV 非核苷酸逆转录酶抑制剂Rilpivirine、Etravirine 及Doravirine 的合成路线进行研究,揭示了许多在高效率有机合成中常用的方法。例如加速反应的微波辐射方法、确定反应最佳参数的正交实验以及利用有机金属催化反应减少反应步骤。在当代的有机化学合成的研究中,不仅需要获得在化学、立体化学上正确的产物,更需要在得到正确产物的基础上提高合成的效率,才能在工业上进行大规模的生产。如何根据化学热力学、化学动力学的原理提升产率和效率成为了接下来合成研究的核心。同时,随着量子化学以及催化剂的发展,一些物理方法以及高效的催化剂成为了在原理上提升有机合成高效性的工具。
猜你喜欢
中国药学药品知识仓库(2022年10期)2022-05-29 02:59:32
云南化工(2021年9期)2021-12-21 07:44:20
汕头大学学报(自然科学版)(2020年4期)2020-12-14 07:04:58
山东农业科学(2019年9期)2019-12-09 01:52:35
中国计划生育学杂志(2017年3期)2017-06-01 12:10:12
现代检验医学杂志(2016年4期)2016-11-15 02:01:14
听力学及言语疾病杂志(2015年5期)2015-12-24 01:47:05
结构化学(2014年5期)2014-12-15 08:56:20
重庆三峡学院学报(2014年3期)2014-07-16 09:37:56
无机化学学报(2014年12期)2014-02-28 17:34:01
