
固相萃取-高效液相色谱法同时测定水产品中15 种磺胺类药物残留
2024-02-27 15:08汤凯洁冀坤霞聂馨梦郎济洲杜华英顾小红
食品与生物技术学报 2024年1期
李 倩, 汤凯洁, 冀坤霞, 聂馨梦, 郎济洲, 杜华英, 顾小红
(1. 江西农业大学食品科学与工程学院, 江西 南昌 330045; 2. 食品科学与资源挖掘全国重点实验室, 江南大学,江苏 无锡 214122)
磺胺类药物 (SAs) 是一类以对氨基苯磺酰胺(sulfanilamide,SN) 为基本结构的合成抗菌药的总称,是我国目前使用量居第二的兽用抗菌药。SAs 具有抗菌谱广、价格低、使用方便等优点,被广泛用于治疗、预防动物疾病或促进动物生长[1]。 SAs 的超剂量、超范围使用导致其在动物体内蓄积,并通过食物链传给消费者[2],引起人体的过敏反应、急慢性毒性,使细菌产生抗药性,甚至有致癌、致畸、致突变等风险[3-4]。 为保障消费者安全,多个国家和政府都制定了SAs 最大残留限量 (maximum residue limit,MRL) 标准。 如国际食品法典委员会(Codex Alimentarius Commission,CAC) 规定动物肌肉、肝脏、肾脏、脂肪等食品中SAs 的MRL 为100 μg/kg[5];我国农业农村部235 号公告规定所有动物源性食品中SAs 总的MRL 为100 μg/kg,磺胺二甲基嘧啶在牛奶中的MRL 为25 μg/kg[6];欧盟地区、日本等国家也对SAs 的MRL 做出了相同规定。 可见SAs在国内外都有严格的限量标准,为加强监管,建立一种准确度好、灵敏度高的SAs 多残留检测方法具有重要意义。根据统计,我国SAs 在2013 年的年消耗量达到了7 890 t, 而其中88.5%是用于养殖业[7]。SAs 在畜禽类(猪肉、鸡肉、鸭肉)[8]、蜂蜜[9]、奶牛粪污[10]中都有检出。 在饮用水[11]、虾[12]以及泰国进口的鲶鱼[13]中都检测出了磺胺嘧啶、磺胺二甲基嘧啶、磺胺甲恶唑与磺胺甲恶嗪。 董燕等对昌吉市3 种食用鱼(草鱼、鲤鱼、鲢鱼)体内磺胺类药物残留进行分析,在20 批次不同季度的鲤鱼体内均检出了SAs[14]。可见水产品中SAs 的残留不容乐观,而江西水产中SAs 残留目前未见报道。目前,检测SAs 残留的主要方法有免疫分析法与仪器分析法。 免疫分析法有酶联免疫吸附法(enzyme-linked immunosorbent assay,ELISA)[15]与胶体金法 (colloidal gold -based immunochromato-graphic assay,CGIA)[16], 这两种方法都是根据抗原与抗体特异性结合进行检测,具有灵敏度高、检测速度快、特异性强等优点。 但目前国内外这种快速检测方法仅用于筛查,筛查到的阳性样品仍然需要采用仪器方法进一步确证。 仪器分析法有气相色谱-串联质谱法 (gas chromatographytandem mass spectrometry,GC-MS/MS)[17-18]、 高效液相色谱法[19-21]、超/高效液相色谱-串联质谱法(ultra/high liquid chromatography tandem mass spectrometry,UPLC/LC-MS/MS)[22-26]等。这些方法操作自动化程度高,定性定量准确,HPLC 仪器相比其他两种仪器价格更便宜,是目前国内多数实验室和检测中心都配备的仪器,因此其应用更广。 由于杂质峰会干扰检测和堵塞色谱柱, 因此使用HPLC 检测前需要对样品进行复杂的净化富集预处理。 食品样品基质复杂,为了去除样品中杂质的干扰,通常都需要采用固相萃取柱净化和富集处理, 如采用MCX 固相萃取柱[27]、PCX 固相萃取柱[28]和HLB 固相萃取柱[29]净化富集鸡肉样品,QuEChERS 净化管[30]处理鱼肉制品等。 这几种净化富集小柱中,PCX 固相萃取柱填料为混合型弱阳离子交换聚合物材料,具有离子交换和反相保留双重作用,其比表面积大,适用的pH范围广,结合容量大,而且价格较MCX 固相萃取柱与HLB 固相萃取柱低50%~70%, 适用于富集和提取碱性化合物,如SAs。 但目前未见采用PCX 固相萃取柱预处理鱼肉样品报道。 以对氨基苯磺酰胺为SAs 母体结构, 在酰胺键上异构不同官能团衍生了30 多种不同的SAs 抗菌药,不同SAs 由于其极性和分子大小不同,其高效液相色谱分离条件不同。 《食品安全国家标准动物性食品中13 种磺胺类药物多残留的测定高效液相色谱法》(GB 29694—2013)[27]报道了13 种SAs 的检测方法, 作者选择了15 种SAs 为研究对象, 其中有7 种是国家标准方法中没有包含的。 研究15 种SAs 的HPLC 分离条件和价格更低廉的PCX 固相萃取柱对水产品中SAs 提取净化条件, 建立了同时测定水产品中15 种SAs 的SPE-HPLC 分析方法。该方法与国家标准方法比较,不仅增加了两种SAs 的检测,且有7 种SAs 与国家标准中不同, 另外筛选的PCX 固相萃取柱价格低,重复和重现性好,方法的检出限和灵敏度能满足国家标准中的限量要求,为水产品中SAs 的残留检测提供参考依据。
1 材料与方法
1.1 试剂与材料
1.1.1 15 种SAs 标准品磺胺嘧啶(sulfadiazine,SD)、 磺胺噻唑 (sulfathiazole,ST)、 磺胺吡啶(sulfapyridine,SPD)、 磺胺甲基嘧啶(sulfamerazine,SM1)、磺胺对甲氧嘧啶(sulfameter,SMD)、磺胺甲噻二唑 (sulfamethizole,SMT)、 磺胺二甲基嘧啶(sulfamethazine,SM2)、磺 胺 甲 氧 哒 嗪(sulfamethoxypyridazine,SMP)、 磺 胺 氯 哒 嗪(sulfachloropyridazine,SCP)、 磺 胺 甲 恶 唑(sulfamethoxazole,SMZ)、 磺 胺 间 甲 氧 嘧 啶(sulfamonomethoxine,SMM)、 磺胺多辛(sulfadoxin,SDO)、磺胺异噁唑(sulfisoxazole,SIZ)、磺胺氯吡嗪钠 (sulfachloropyrazine sodium,SPZ)、 磺胺喹噁啉(sulfachinoxalin,SQ): 北京世纪奥科生物技术有限公司。
1.1.2 其他试剂与材料甲醇、 乙腈 (均为色谱纯):美国TEDIA 天地试剂公司;冰乙酸、乙酸乙酯、正己烷(均为色谱纯)、无水硫酸钠(分析纯):美国Macklin 公司;十二水合磷酸氢二钠、氨水(均为分析纯): 西陇科学股份有限公司; 无水磷酸二氢钠(分析纯): 阿拉丁生化科技股份有限公司;QuEChERS 净化管(15 mL,含有吸附剂PSA 0.15 g、C180.15 g、MgSO40.9 g): 逗点生物科技有限公司;PCX 固相萃取柱(60 mg,3 mL):博纳艾杰尔科技有限公司;HLB 固相萃取柱(60 mg,3 mL):德国CNW公司;实验用水为超纯水。 草鱼:购于当地农贸市场。
1.2 仪器与设备
Waters E2695 高效液相色谱仪 ( 配有Waters2998 光电二极管阵列检测器 (PDA)和Empower 色谱工作站)、Waters C18(4.6 mm×250 mm,5.0 μm)色谱柱:美国Waters 公司;HC-2518R 型高速冷冻离心机: 安徽中科中佳科学仪器有限公司;HSE-24B 真空固相萃取装置:上海皓庄仪器有限公司;WD-12 氮吹仪: 杭州奥盛仪器有限公司;Milli-Q 纯水仪:德国Millipore 公司。
1.3 实验方法
1.3.1 色谱条件Waters C18(4.6 mm×250 mm,5.0 μm) 色谱柱;柱温35 ℃;进样体积10 μL;流量1 mL/min;PDA 波长270 nm;流动相:溶液A 为甲醇, 溶液B 为含体积分数1.1%乙酸的PBS 缓冲液(0.01 mol/L); 梯度洗脱程序:0~30 min, 体积分数95% 溶液B;30~50 min, 体积分数80%溶液B;50~55 min,体积分数60%溶液B;55~60 min,体积分数60%溶液B;60~65 min,体积分数95%溶液B。
1.3.2 标准溶液与流动相的配制分别准确称取15 种SAs 标准品15.0 mg 于50 mL 容量瓶中,用甲醇溶解定容,配制成质量浓度为300 μg/mL 的单标储备液。 再由300 μg/mL 的单标储备液配制成质量浓度为10.0 μg/mL 的单标和系列质量浓度混合标准溶液(0、0.5、1.0、5.0、8.0、10.0 μg/mL),于-18 ℃下避光密闭保存。
1.3.3 流动相的配制含体积分数1.1%乙酸的PBS 缓冲液(0.01 mol/L):准确称取3.58 g Na2HPO4、1.56 g NaH2PO4, 用超纯水溶解, 加11 mL 的冰醋酸,超纯水定容至1 L。
1.3.4 样品的前处理
1) 提取 参考国家标准 《水产品抽样规范》(GB/T 30891—2014)[31],取肌肉、鱼皮等可食部分搅碎混匀备用,称取匀浆试样1.00 g(精确至0.01 g),置于50 mL 已加有5.00 g 无水硫酸钠的聚丙烯具塞离心管中,加入3 mL 的乙酸乙酯提取溶剂,涡旋混匀2 min, 超声提取30 min,8 000 r/min 离心5 min,将上清液转移至另一离心管中,残渣再用3 mL的乙酸乙酯重复提取一次,合并两次提取液。
2) 净化与过柱 将乙酸乙酯提取液在45 ℃氮气下吹干, 加入5 mL 0.1 mol/L HCl 和10 mL 正己烷, 涡旋混匀1~2 min, 然后以8 000 r/min 离心5 min,弃去上层正己烷,下层的盐酸层留待PCX 固相萃取柱净化。
PCX 固相萃取柱洗脱程序:PCX 固相萃取柱依次用3 mL 甲醇、3 mL 0.1 mol/L HCl 活化, 将离心所得的盐酸层样液以5 滴/s 的速度过PCX 固相萃取柱,再用3 mL 甲醇、3 mL 0.1 mol/L HCl 淋洗,最后用4 mL 体积分数5%氨化甲醇洗脱。HLB 固相萃取柱洗脱程序:HLB 固相萃取柱依次用3 mL 甲醇、3 mL 超纯水活化, 将离心所得的盐酸层样液以5滴/s 的速度过HLB 固相萃取柱, 依次用3 mL 水、3 mL 体积分数5%甲醇淋洗, 最后用4 mL 体积分数5%氨化甲醇洗脱。 QuEChERS 净化管应用程序:将离心所得的盐酸层样液转移到QuEChERS 净化管中,涡旋混匀1 min,8 000 r/min 离心5 min,取上清液备用。 分别收集PCX 固相萃取柱、HLB 固相萃取柱的洗脱液与QuEChERS 净化管中净化后的上清液,于45 ℃氮气吹干,用1 mL 初始比例流动相复溶,复溶液过0.22 μm 滤膜,留待高效液相色谱测定。
1.3.5 数据处理所有的实验至少重复进行3 次,不同提取溶剂的提取回收率显著性分析采用IBM SPSS Statistics 27 处理,P<0.05 表示差异显著;HPLC 数据采用Empower 色谱工作站采集; 使用Origin 2018 和Excel 2016 进行图表的绘制。
2 结果与分析
2.1 15 种SAs 紫外吸收波长的确定
采用高效液相色谱在200~400 nm 波长进行扫描,确定了15 种SAs 的最大吸收波长,结果见表1。由表1 可知,所检测的15 种SAs 在250~285 nm 时有较强的吸收, 后续实验选择270 nm 作为15 种SAs 的最大吸收波长,即检测波长。

表1 15 种SAs 的最大吸收波长Table 1 Maximum absorption wavelengths of 15 kinds of SAs
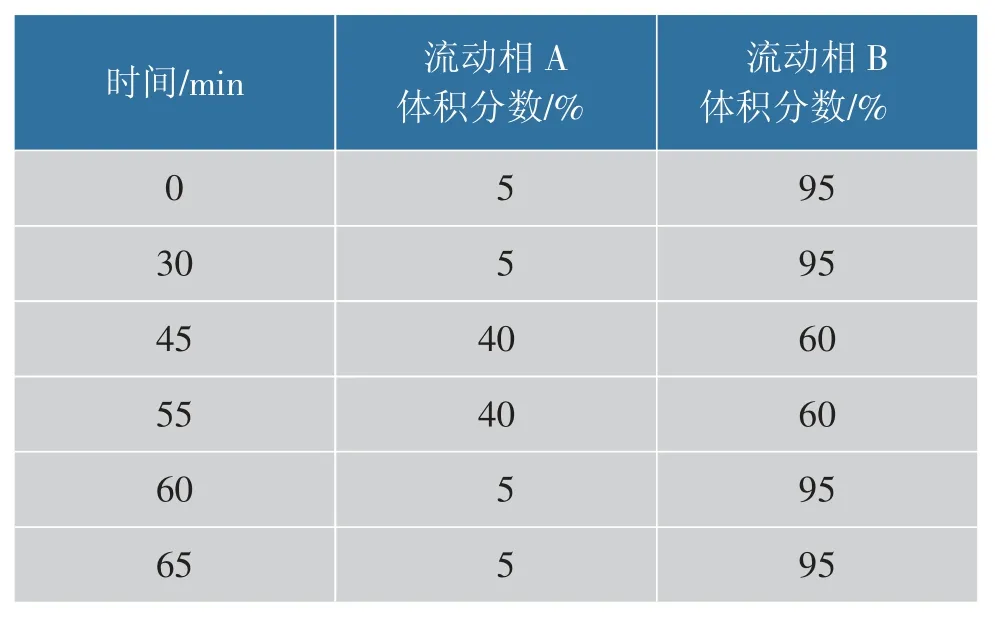
表2 流动相梯度洗脱程序1Table 2 Mobile phase gradient elution procedure 1

表3 流动相梯度洗脱程序2Table 3 Mobile phase gradient elution procedure 2

表4 流动相梯度洗脱程序3Table 4 Mobile phase gradient elution procedure 3
2.2 流动相与梯度洗脱程序优化
为了实现15 种SAs 完全分离, 流动相条件采用梯度洗脱,实验重复3 次。 实验设计了3 种不同的梯度洗脱分离程序, 考察不同梯度洗脱对15 种SAs(各组分质量浓度均为2 μg/mL)分离效果的影响, 梯度洗脱程序1、2、3 分别如表1、2、3 所示,分离色谱图如图1 所示。 采用梯度洗脱程序1 分离得到的色谱图如图1 中A 所示, 在15~35 min,SD、ST、SPD、SM1 四种SAs 完全分离, 但响应较低;在40~47 min,9 种SAs 响应较好,但未实现完全分离。采用梯度洗脱程序2 分离得到的色谱图如图1 中B所示,绝大部分SAs 的出峰时间提前,13~37 min 色谱峰完全分离, 但SAs 响应值较低;SDO 与SIZ 未实现完全分离。 采用梯度洗脱程序3 分离得到的色谱图如图1 中C 所示,色谱图SMD(峰5)与SMT(峰6) 两峰之间的分离度为1.2, 实现了98%的分离,其余相邻两峰之间分离度均不小于1.5,实现了完全分离,且峰形对称,更尖锐。 因此,后续实验选择梯度洗脱程序3 进行分析与定量。

图1 15 种SAs 3 种梯度洗脱程序高效液相色谱图Fig. 1 High performance liquid chromatograms of 15 kinds of SAs under 3 kinds of elution procedures
2.3 SAs 标准曲线的绘制
将1.3.2 中所配制的系列质量浓度混合标准溶液依次进行高效液相色谱测定,以溶液质量浓度为横坐标(x),峰面积为纵坐标(y),绘制SAs 的标准曲线,15 种SAs 的标准曲线如图2 所示。
2.4 提取条件的优化
2.4.1 样品提取溶剂的选择15 种SAs 的衍生基团不同,其极性大小不同。 不同的提取溶剂会影响其提取回收率,一般采用极性有机溶剂进行提取以避免提取出过多的脂肪。 根据15 种SAs 的理化性质和相关文献,对比了体积分数3%乙酸乙腈、乙酸乙酯、体积分数80%乙醇、乙腈、体积分数80%乙腈与二氯甲烷6 种提取溶剂对15 种SAs 提取效果(n=6)。 在空白鱼肉样品中加入1 mL 5 μg/g SAs 混合标准溶液,按照1.3.4 中的提取方法处理,计算不同提取溶剂的提取回收率(n=6),比较结果如图3所示, 此外, 采用新复极差法 (shortest significant ranges,SSR) 比较不同提取溶剂的提取回收率是否具有显著差异,结果如表5 所示。 由图3 与表5 可见,采用乙酸乙酯提取时杂峰干扰小,对15 种SAs的提取回收率在84.1%~102.7%, 这与郑斌等运用高效液相色谱测定水产品中的14 种SAs (88.9%~98.6%)[32]结果一致且提取回收率略高;乙腈的提取回收率在64.6%~92.0%,其对极性弱的SAs 提取效果不佳,尤其对SIZ 与SPZ,提取回收率仅为65.0%左右,而且在用正己烷净化过程中会产生乳化贴壁现象,且色谱图上杂峰较多,杂质干扰大;体积分数3%乙酸乙腈与二氯甲烷提取回收率在70.8%~99.0%,其对弱极性SAs 提取效果较好,而对ST 与SPZ 提取回收率较低;体积分数80%乙醇和体积分数80%乙腈杂质干扰相对较小, 对15 种SAs 的提取回收率在68.3%~94.9%。 由表5 可见,因检测的SAs 和所比较的提取溶剂种类较多,6 种不同的提取溶剂间对每种磺胺类药物提取回收率间差异的显著性均不完全一致, 但乙酸乙酯对大部分的SAs提取回收率与其他提取溶剂相比存在显著差异(P<0.05),因此,综合考虑,最终选择乙酸乙酯作为提取溶剂。

图3 不同提取溶剂对15 种SAs 的提取回收率比较Fig. 3 Comparison of extraction recoveries of 15 kinds of SAs using different solvents
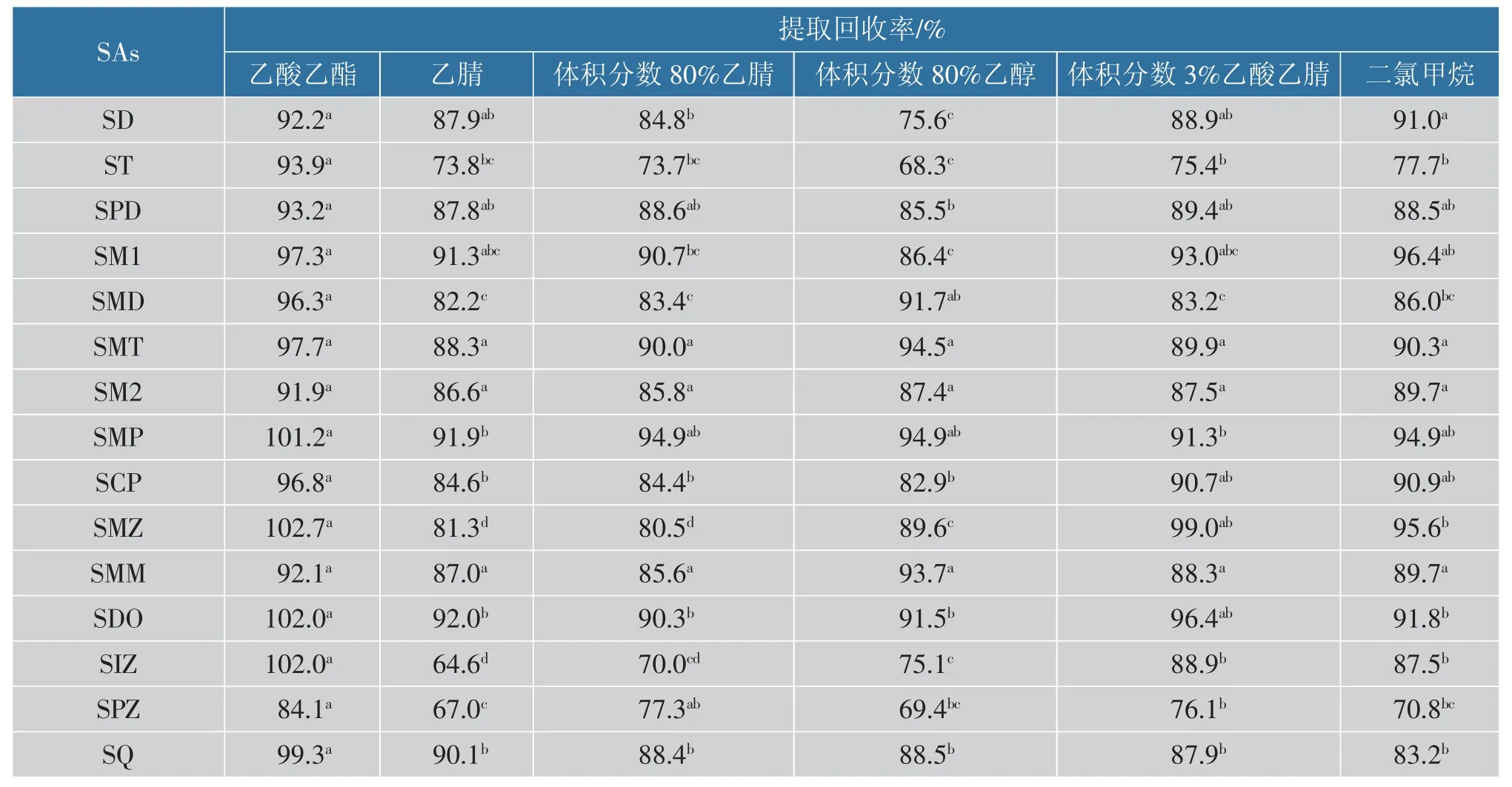
表5 6 种不同提取溶剂对15 种SAs 提取回收率的差异显著性分析(SSR0.05)Table 5 Significance analysis of the differences in the extraction recoveries of 15 kinds of SAs with 6 different extraction solvents (SSR0.05)
2.4.2 净化小柱的筛选样品按照1.3.4 中的提取与正己烷脱脂,下层的HCl 层样液分别过PCX 固相萃取柱、HLB 固相萃取柱与QuEChERS 净化管,对SAs 的加标回收率进行比较。
结果发现PCX 固相萃取柱净化效果好,杂质干扰少,加标回收率在72.1%~100.2%,略高于其他学者[33-34]用PCX 固相萃取柱对猪肉(70.2%~89.9%)与蜂蜜(77%~99%)样品中SAs 检测的加标回收率;虽然HLB 固相萃取柱净化后杂质干扰也少,但加标回收率低, 仅为6%~10%, 说明HLB 固相萃取柱对SAs 吸附能力差;QuEChERS 净化管净化后杂质干扰最大,且加标回收率低,为9%~17%。 因此,选择PCX 固相萃取柱进行样品净化。
2.4.3 上样溶剂的筛选实验研究发现,不同极性的上样溶剂会影响PCX 固相萃取柱对SAs 的保留效果。 为了最大程度地保留SAs,提高加标回收率,溶解样品的溶液极性必须较弱。 实验分别对体积分数2%乙酸水溶液(pH 3)、 质量分数0.05% NaOH溶液(pH 7)、PBS 缓冲液、0.1 mol/L HCl 4 种上样溶剂的SAs 吸附效果进行了比较。 样品按照1.3.4 中的方法提取后, 分别加入5 mL 上述的4 种上样溶剂与10 mL 正己烷,净化后弃去正己烷层过PCX 固相萃取柱后供HPLC 分析,结果如图4 所示。由图可见,以0.1 mol/L HCl 为上样溶剂时,检测上样流出液几乎未检测到15 种SAs 的色谱峰, 说明15 种SAs 基本被PCX 固相萃取柱保留(见图4 中A),加标回收率为73.1%~109.6%; 以PBS 缓冲液为上样溶剂时, 检测上样流出液也几乎未检测到15 种SAs,但在5~7 min 处杂峰干扰较多(见图4 中B),加标回收率为68.7%~97.5%; 而体积分数2%乙酸水溶液 (pH 3) 和质量分数0.05% NaOH 溶液(pH 7)为上样溶剂时,在上样流出液中检测到多种明显的SAs 色谱峰, 两者对15 种SAs 的加标回收率偏低,分别为56.6%~84.2%与9.0%~47.6%。 尤其是质量分数0.05% NaOH 溶液(pH 7)为上样溶剂时,流出液中有6 种SAs 色谱峰很高,说明采用这两种溶剂上样时对绝大部分的SAs 保留效果较差,可能是SAs 易溶于稀碱溶液,导致加标回收率降低。

图4 不同上样溶液对15 种SAs 的保留效果Fig. 4 Effects of different loading solutions on the retention of 15 kinds of SAs
2.4.4 淋洗条件的选择淋洗是为了洗掉干扰组分,而SAs 被保留。 分别比较了3 mL 的水、体积分数2%甲酸水溶液与0.1 mol/L HCl 淋洗PCX 固相萃取柱,分别收集不同淋洗液后采用高效液相色谱检测分析,发现均未检测到15 种SAs,说明三者淋洗液极性强度适中,都可以淋洗杂质。 后续实验选择3 mL 0.1 mol/L HCl 作为淋洗液。
2.4.5 洗脱条件的选择为了完全洗脱PCX 固相萃取柱吸附的15 种SAs,根据相关文献报道,本文中比较了3 种洗脱液(甲醇-乙酸乙酯-氨水(体积比为49∶49∶2) 混合液[35]、甲醇[36]、体积分数5%氨化甲醇[37])对SAs 的洗脱效果,结果如图5 所示。 当以体积分数5%氨化甲醇洗脱时,样品中15 种SAs 几乎完全被洗脱,洗脱回收率在78.7%~89.3%(见图5中A);以甲醇-乙酸乙酯-氨水(体积比为49∶49∶2)混合液洗脱时,除SPD 未被洗脱,其余SAs 均被洗脱,洗脱回收率略低于体积分数5%氨化甲醇(见图5 中B);以甲醇洗脱时,只有少部分的目标物能被洗脱, 且洗脱回收率仅为7.0%~26.0%(见图5 中C)。因此,强极性的体积分数5%氨化甲醇洗脱效果最好。 此外, 比较了4 种不同体积分数 (1%、3%、5%、7%)的氨化甲醇对15 种SAs 的洗脱效果。结果显示随着氨化甲醇体积分数的增加,洗脱回收率呈上升趋势。 这与王宏宇等采用SPE-UPLC 同时测定猪肉中22 种磺胺类药物残留所优化的洗脱条件[38]一致。 体积分数为5%和7%的氨化甲醇对15 种SAs 的洗脱回收率分别为78.7%~89.3%、82.5%~103.5%,两者洗脱回收率比较接近。体积分数1%氨化甲醇洗脱回收率仅为21.4%~45.8%,而体积分数3%氨化甲醇为62.1%~82.3%。最终选择以体积分数5%氨化甲醇作为PCX 固相萃取柱的洗脱液。

图5 不同洗脱液对15 种SAs 的洗脱效果Fig. 5 Elution effects of different eluents on 15 kinds of SAs
2.5 加标回收率与精密度
准确称取1.00 g 空白鱼肉样品,进行3 个质量分数水平(500、1 000、2 000 μg/kg) 的加标回收实验; 按照1.3.4 的前处理方法对样品进行处理后采用高效液相色谱检测。 每个水平重复实验6 次,使用标准曲线校正并测定SAs 质量分数,计算加标回收率、日内与日间的RSDs,空白鱼肉样品与加标鱼肉样品色谱图见图6 中A 和B,其结果见表6。由表6 可见, 除2 000 μg/kg SPZ 的加标回收率略低(73.1%), 其他SAs 在3 个质量分数下的加标回收率在78.6%~109.6%,RSDs 在1.0%~6.7%,日内与日间的RSDs 分别在1.3%~6.7% 与1.0%~6.4%,说明所建立方法准确,重复性与重现性均较好。
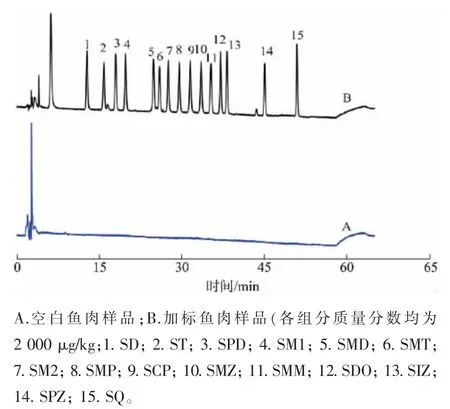
图6 空白鱼肉样品及加标样品的高效液相色谱图Fig. 6 High performance liquid chromatograms of the blank and spiked fish samples

表6 15 种SAs 的加标回收率与精密度Table 6 Recoveries and precision of 15 kinds of SAs
2.6 检出限与定量限
准确取1.00 g 的空白草鱼样品,并向其中添加不同质量浓度的混合标准溶液, 按照1.3.4 的前处理方法对样品进行处理后上机检测。 按照相关文献所报道的关于分析方法验证的指导原则[39-40],用基于响应值的标准偏差和标准曲线的斜率来定义方法的检出限 (limits of detection,LODs) 和定量限(limits of quantitation,LOQs),结果见表7。方法的检出限与定量限分别在10~20 μg/kg 与20~60 μg/kg,该方法的检出限和定量限能满足标准的需要。

表7 该方法的线性范围、线性方程、检出限与定量限Table 7 Linear range, linear equation, LOD, and LOQ of the method
2.7 实际样品的测定
采用实验优化的方法,对从当地市场采购的鲫鱼、鲈鱼、巴沙鱼、黑鱼、虾、草鱼、鲶鱼、鲢鱼、鳙鱼与鳜鱼10 种不同水产品进行了检测, 每种样品重复检测5 次,采用保留时间定性的方式对样品中的SAs 进行筛查, 均未检测出阳性样品,10 种不同水产品HPLC 色谱图如图7 所示, 可见江西水产品中SAs 残留情况并不严重。

图7 10种样品高效液相色谱图Fig. 7 High performance liquid chromatograms of 10 samples
3 结 语
实验优化了15 种SAs 的HPLC 检测条件,筛选了适用于水产品富集与净化的PCX 固相萃取柱,采用高效液相色谱法结合SPE 技术,建立了一种同时测定水产品中15 种SAs 残留的方法。 样品经乙酸乙酯提取,正己烷脱脂,PCX 固相萃取柱净化后,采用Waters C18色谱柱分离检测, 除2 000 μg/kg SPZ 的加标回收率略低(73.1%),其他SAs 在3 个质量分数水平下的加标回收率在78.6%~109.6%,RSDs 在1.0%~6.7%,方法的检出限与定量限分别在10~20 μg/kg 与20~60 μg/kg。采用该方法对江西10种水产品中的15 种SAs 残留进行了检测, 结果均未检出,可见江西水产品SAs 残留情况并不严重。
猜你喜欢
云南化工(2021年11期)2022-01-12
中国油脂(2020年3期)2020-04-10
当代水产(2019年3期)2019-05-14
Coco薇(2017年7期)2017-07-21
无机化学学报(2016年8期)2016-12-06
金色年华(2016年23期)2016-06-15
化学分析计量(2016年1期)2016-03-14
分析测试学报(2015年3期)2016-01-13
分析测试学报(2015年9期)2015-12-17
应用化工(2014年12期)2014-08-16
