
1起ST19型鼠伤寒沙门氏菌引起的食物中毒调查、溯源分析与探讨
2024-02-23 01:57余树坤谭雅心崔紫妍徐兴宇陶志阳
中国人兽共患病学报 2024年1期
余树坤,刘 浪,谭雅心,崔紫妍,徐兴宇,陶志阳
沙门氏菌作为食源性腹泻常见的病原菌之一[1],广泛地存在于自然界中,其传播主要与受污染的动物肉类和蛋类食品有关,能引起各种家禽和哺乳动物的传染病,也可引起人类感染,以肠炎沙门氏菌、鼠伤寒沙门氏菌和德尔卑沙门氏菌感染常见,具有重要的公共卫生意义[2-3]。
2022年1月19日,武汉市某区人民医院发热门诊先后收治多名进食晚餐后出现发热、腹泻、呕吐等胃肠道症状的患者,均来自同一项目工地,具有共同进餐史,调查显示项目工地20名员工于1月19日共同进食晚餐后,其中有7人发病,临床症状以发热、腹泻为主。根据流行病学调查及实验室结果,高度怀疑为沙门氏菌引起的食物中毒。本研究对采集的样本进行了微生物相关致病菌检测,并对检测到的沙门氏菌应用脉冲场凝胶电泳技术进行分子分型和全基因组测序,以期为今后类似事件的调查检测提供参考。
1 材料与方法
1.1 材 料
1.1.1 样本来源 现场共采集相关样本20份,其中7份病例(含2名厨师)肛拭子样本;2份病例粪便样本;5份共同进餐工作人员肛拭子样本;6份外环境涂抹样本来自圆砧板、小砧板、刀把手、灶台。因项目工地食堂无剩余熟食,且无剩余未加工的原材料,无法进行可疑食物样品采集。
1.1.2 主要仪器 高通量测序系统(Illumina公司,型号:Illumina MiSeq),实时荧光定量PCR仪(赛默飞世尔科技有限公司,型号:QuantStudio 7 Flex),全自动核酸提取仪(西安天隆科技有限公司,型号:NP968-C),全自动微生物鉴定药敏分析仪(碧迪医疗器械有限公司,型号:BD Phoenix M50),脉冲场凝胶电泳系统(Bio-Rad公司,型号:Chef mapper),病原微生物生信分析平台(杭州柏熠科技有限公司,版本V5.0)。
1.1.3 培养基与试剂 18种食源性病原体核酸多重实时荧光PCR检测试剂盒(上海卓成惠生,批号:T202207158),SC增菌液(北京路桥有限公司,批号:221216),沙门显色琼脂培养基(青岛海博有限公司,批号:20230227),革兰氏阴性菌鉴定/药敏板(碧迪医疗器械(上海)有限公司,批号:2067245),沙门氏菌属139种诊断血清(宁波天润,批号:20220701), 细菌基因组DNA提取试剂盒(天根生化科技有限公司,批号:X1017),MiSeq Reagent Kit v2 (300-cycles)测序试剂盒(Illumina公司,批号:20680866)。
1.2 方 法
1.2.1 病例定义 自1月17日以来在该工地食堂就餐后出现发热(≥37.3 ℃)、腹泻(≥3次/24 h,并伴大便性状改变)、呕吐等症状,判定为疑似病例。
1.2.2 流行病学调查 按照《食品安全事故流行病学调查技术指南(2021版)》,根据病例定义对搜索到的全部病例进行个案调查。调查内容主要有人口学信息、发病和治疗经过。调查该项目部食堂中所用食物原材料来源,食物加工方法、制作过程,食物原材料及成品保存方法、保存环境等。
1.2.3 致病原的多重PCR检测及分离鉴定
1.2.3.1 样本总DNA/RNA的提取及多重PCR检测 将采集的20份样本充分震荡混匀后,用全自动核酸提取仪提取核酸,按照18种食源性病原体核酸多重实时荧光PCR检测试剂盒操作说明开展多重PCR检测。
1.2.3.2 致病菌的分离鉴定 根据多重PCR初筛结果,将盐水管采集的肛拭子和外环境涂抹样本及粪便样本各吸取少许液体接种到SC增菌液中增菌培养,增菌液划线接种到相应的选择培养基,36 ℃培养24 h后观察结果。可疑菌落分纯培养后采用全自动微生物分析仪进行系统生物鉴定,阳性菌株进行血清学实验。
1.2.4 药敏试验 选用全自动细菌生化鉴定仪配套的革兰阴性细菌药敏卡,包含20种抗生素,对12株菌株进行药敏实验,按照试剂盒说明书操作并判定结果。
1.2.5 PFGE分子分型 使用Xbal限制性内切酶进行内切消化,并按照《国家致病菌识别网实验室监测手册(2022版)》中沙门菌PFGE分型程序操作说明进行菌株分型。经GelRed染色成像,使用BioNumerics软件以90%相似度进行聚类分型。
1.2.6 全基因组测序分析
1.2.6.1 测序 根据细菌基因组DNA提取试剂盒说明书提取DNA,DNA浓度高于100 ng/μL后,经Illumia测序平台测序,然后通过柏熠病原微生物分析平台(V 5.0)用fastp软件(v0.23.2版本)进行数据质量质控,过滤后的clean data使用SPAdes软件(v3.15.3版本)对沙门氏菌全基因组序列进行组装。
1.2.6.2 生物信息学分析 根据沙门氏菌的基因组信息和抗性基因数据库中的基因信息,使用abricate(V1.0.1)软件进行耐药注释分析 。沙门氏菌血清型鉴定使用seqsero2(V1.2.1)软件分析注释。使用mlst(V2.23.0)针对沙门氏菌管家基因aroC、dnaN、hemD、hisD、purE、sucA、thrA进行多位点序列分型(MLST),使用cgmlstfinder(V1.2.0)进行核心基因组多位点序列分型(cgMLST)。使用snippy对样本进行全基因组单核苷酸位点分析(wgSNP)。对比NCBI assembly同源数据库,采用fastANI(V 1.33)对数据库进行同源性分析,选取同源性排序前50株样本使用fasttree(V2.1.11)构建进化树,计算进化树上进化距离关系,从而获得最近源序列。
2 结 果
2.1 流行病学调查结果 该项目工地共有员工20名,可提供三餐及住宿,食堂由2名厨师(肖某、陈某)轮流值班,环境卫生一般,宿舍为搭建的活动板房,无空调,密封条件差。经调查,符合患者定义的共有7例,首例病例发病时间为1月20日0∶00,末例病例发病时间为1月20日6∶00,发病高峰为1月20日0∶00-02∶00。最短潜伏期约为7 h,最长潜伏期约13 h,平均潜伏期为9.1 h。7例病例临床症状以发热、呕吐、腹泻为主,并伴有头痛、头晕等症状。患者均为成年男性,最小32岁,最大71岁。所有患者经抗炎补液及对症治疗后均已痊愈返岗。
2.2 样本检测结果
2.2.1 多重PCR鉴定结果 20份样本中,12份样本多重初筛沙门氏菌阳性,其中病例肛拭子样本7份、病例粪便样本2份,工作人员肛拭子样本3份,其余样本均为阴性。
2.2.2 沙门氏菌分离鉴定及血清学结果 参考多重PCR结果,对12份样本进行沙门氏菌分离,增菌后的样本中均检出疑似沙门氏菌,经革兰染色、生化系统鉴定均为阳性,与多重PCR结果一致,菌株编号为DXH001~DXH012,其中DXH010与DXH005为病例5不同发病时期样本分离株,DXH011与DXH002为病例2不同类型样本分离株,DXH012与DXH003为病例3不同类型样本分离株。
对分离到的12株沙门氏菌进行血清型分析,A-F沙门菌多价血清凝集阳性,O4、O12抗原血清凝集阳性,Hi、H2抗原血清凝集阳性,生理盐水对照阴性。得到的抗原为4,12:i:2,查询Kauffman-White表,均为鼠伤寒沙门氏血清型,见表1。

表1 食物中毒相关样本分离结果Tab.1 Isolated Salmonella strains from samples related to food poisoning
2.3 药敏分析结果 12株菌株耐药谱完全一致,均对阿米卡星、氨苄西林、头孢唑啉、庆大霉素、哌拉西林、 四环素耐药,其余14种均敏感。
2.4 PFGE分析结果 考虑DXH010与DXH005(病例5)、DXH011与DXH002(病例2)、DXH012与DXH003(病例3)分别来自同一人员不同时期或不同类型样本分离株,且其生化和血清分型结果一致,后续PFGE和基因测序仅选取DXH001~DXH009菌株进行分析。
将上述9株沙门氏菌株使用限制性内切酶XbaI酶切后进行PFGE,胶块经Gelred染液染色后成像,结果显示9株沙门氏菌产生的电泳条带均相同且分子量大小一致,再将9株沙门氏菌的脉冲场电泳图谱导入BIoNumercis软件进行聚类分析,结果显示9株菌株相识度为100%,说明为同一型别菌株,见图1。

图1 9株沙门氏菌分子分型聚类分析图Fig.1 Diagram of molecular typing and clustering analysis of nine strains of Salmonella
2.5 基因测序与溯源分析结果
2.5.1 全基因组测序、组装结果 经Illumina测序平台和fastp软件质控后,共得到约4.2 G的原始数据,组装后的9株沙门氏菌的基因组大小在4 929 134~4 930 589 bp,GC含量为52.15%~52.16%。
2.5.2 血清凝集分型与全基因组分型结果比较 WGS分型结果均为B群鼠伤寒沙门氏菌,O1、O4、O15、O12抗原血清凝集阳性,Hi、H1、H2抗原血清凝集阳性,与常规血清分型结果一致,两种方法符合率为100%。
2.5.3 耐药基因预测分析与耐药表型比较 基因组耐药基因注释分析发现,每株鼠伤寒沙门氏菌含有10种数量相等的耐药基因,作用机制全部为抗生素外排型。预测显示,9株菌株基因组均含有单菌霉碳、碳青霉烯、头孢菌素、头霉素、青霉烷、利胆醇类、青霉烯、氟喹诺酮、甘氨酰环素、四环素、利福霉素、三氯生、氨基香豆素、硝基咪唑、氨基糖苷类耐药基因。进一步分析发现多种耐药基因可针对一种抗生素产生抗性,例如预测3种耐药基因acrB、mdsB和mdsC均可产生对青霉烷类、氟喹诺酮、氨基香豆素耐药,mdsB和mdsC耐药基因同时可以产生头孢菌素耐药。通过比较,WGS预测的沙门氏菌耐药基因与药敏结果的符合率较高,对氨基糖苷类、头孢菌素、青霉烷、四环素预测的结果完全一致,见表2。

表2 9株鼠伤寒沙门氏菌基因组耐药基因注释分析结果Tab.2 Genome drug resistance gene annotation analysis of nine strains of Salmonella typhimurium
2.5.4 MLST与cgMLST分型结果 为了进一步对9株沙门氏菌进行基因组溯源分析,我们首先使用MLST分析菌株间的相关性。基于全基因组序列的MLST分析结果显示9株菌株均为ST19型别。考虑到MLST分辨力低,对不同暴发或者没有直接流行病学关联的菌株区分能力差,我们进一步进行核心基因组多位点序列分型(core genomemultilocus sequence typing,cgMLST)分析,cgMLST 检测和比对成百上千个基因位点的序列差异,比传统的 MLST 方法具有更高的分辨力。分析结果显示,其分型结果均为cgST 264803 型别,见表3。

表3 cgMLST结果统计信息表Tab.3 The cgMLST results of statistical information
2.5.5 核心基因组单核苷酸多态性分析结果 考虑到同一起暴发性事件往往为同一传播链样本,其基因组差异相对较小。全基因组测序的单核酸多态性分型(whole genome-based singlenucleotide polymorphisms,wgSNP) 是在全基因组序列的水平上选择一定数目的 SNP,比较不同细菌基因组中 SNP 的信息,具有更高的分辨率。9株鼠伤寒沙门氏菌间存在有17个变异位点,样本之间直接SNP差异数为1~6个位点。9株菌SNP最小生成树见图2。
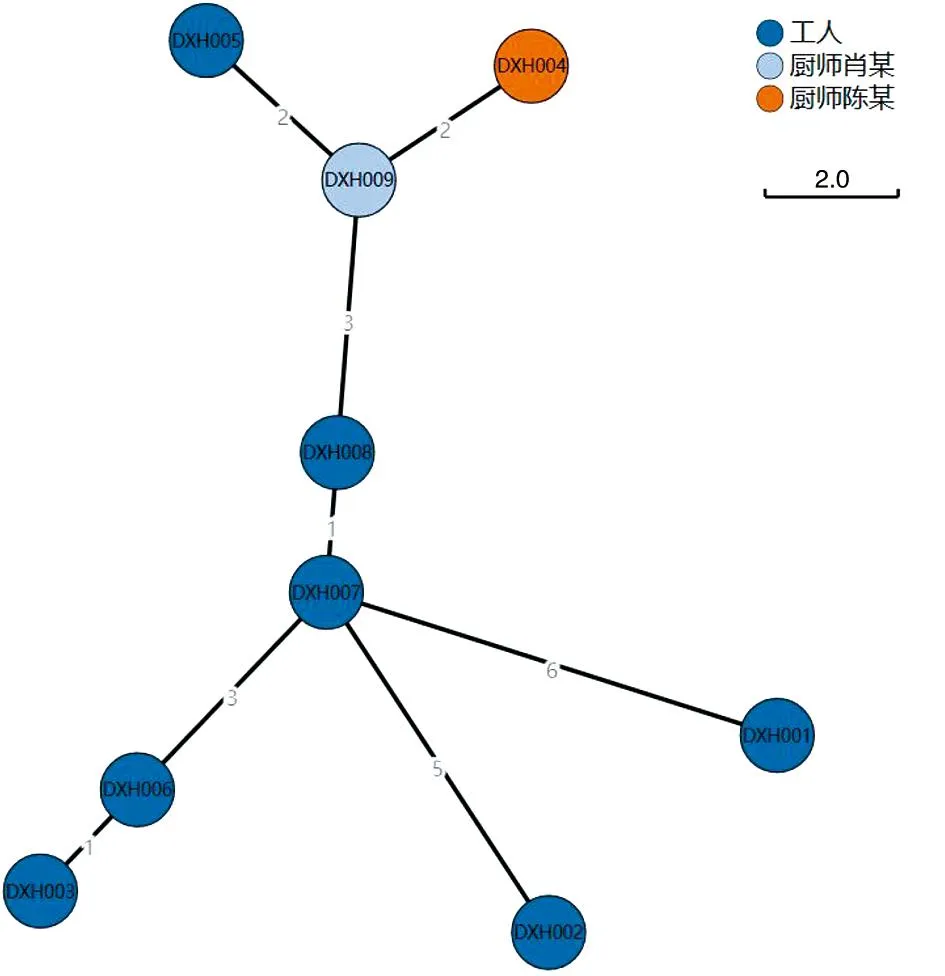
图2 9株ST19型的鼠伤寒沙门氏菌最小生成树分析图Fig.2 Diagrams of minimum spanning trees for nine strains of Salmonella typhimurium ST19
2.5.6 系统发育分析 为了更全面的分析样本之间的系统进化关系,我们加入NCBI (https://www.ncbi.nlm.nih.gov/)数据库中筛选过滤后的完整基因组中沙门氏菌1 188株的数据,然后进行ANI同源性分析,选取同源性排序前50株样本使用fasttree构建进化树。9例样本之间同源性明显高于来源于数据库中的其他样本,并且处于同一进化分支上。在与公共数据库中其他样本比较时,我们发现同源性最高的是来源于江西的GCF_020221795.1样本,详见图3。
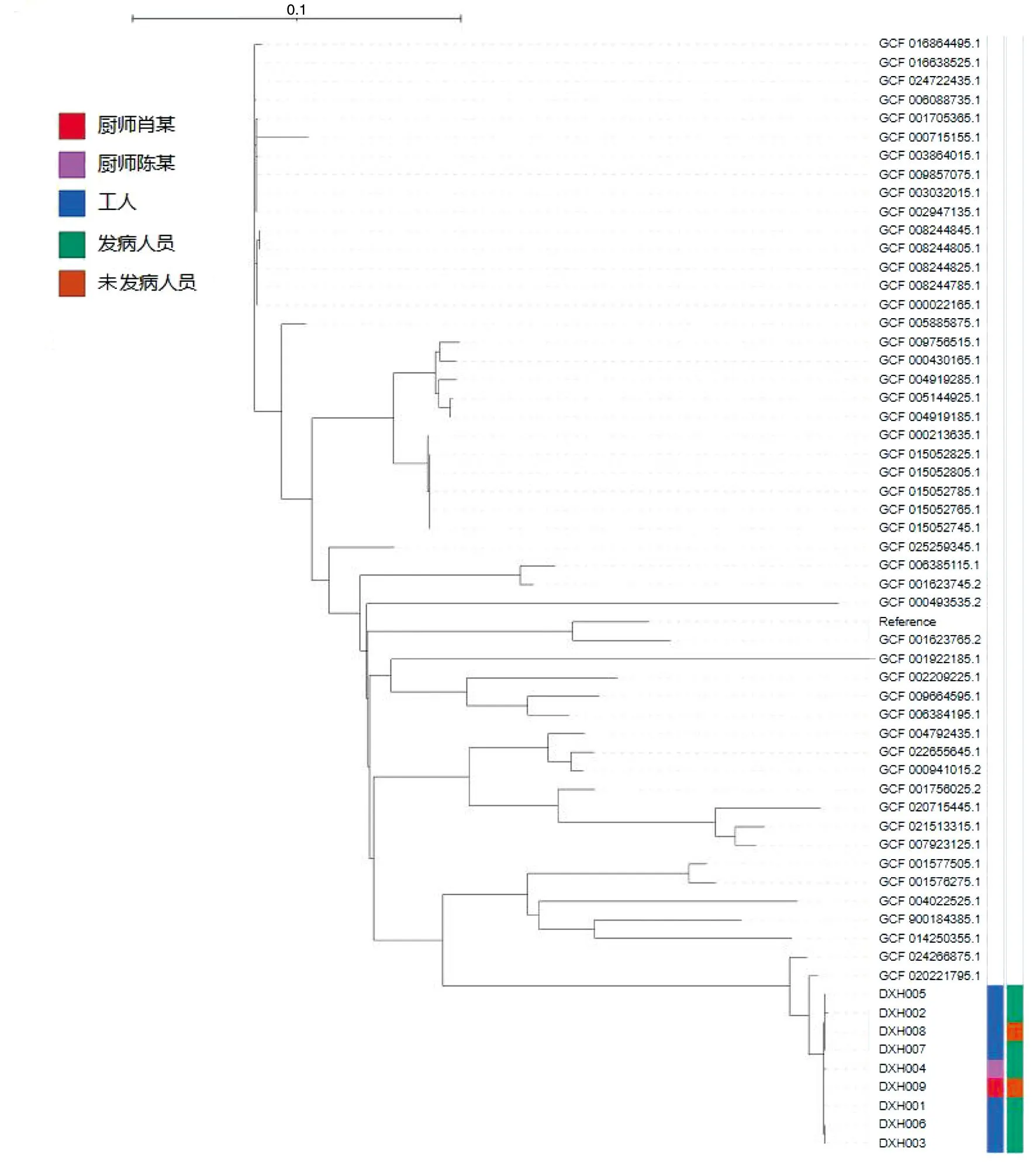
图3 59株沙门氏菌(9株ST19型的鼠伤寒沙门氏菌与50株NCBI数据库沙门氏菌)进化树与样本来源分析图Fig.3 Analysis of the evolutionary tree and sample sources of 59 strains of Salmonella, including nine strains of Salmonella typhimurium ST19 and 50 strains of Salmonella obtained from the NCBI database
3 讨 论
沙门氏菌属是一类危害人和动物健康的重要致病菌,广泛地存在于自然界中,其传播主要与受污染的动物肉类和蛋类食品有关。在世界各国的各类细菌性食物事件中,沙门氏菌引起的食物中毒事件数量常居榜首,其中以鼠伤寒沙门氏和肠炎沙门氏菌感染最常见。沙门氏菌引起的食物中毒的发病潜伏期为6~48 h,患者主要症状包括不同程度的恶心、呕吐、发热、腹痛、腹泻,严重者可出现身体脱水和电解质紊乱[4],本次食物中毒事件中的患者符合这一发病特征,患者在同一时间、同一地点用餐后发病,临床症状相似,发病潜伏期7~13 h,流行曲线呈点源式暴发。根据流行病学调查以及实验室检测结果判断为一起由鼠伤寒沙门氏菌感染引起的食物中毒事件。鼠伤寒沙门氏菌病一年四季均可发生,但主要发生在第二季度和第三季度,没有严格的地域性。
本次事件发生当天气温为1~13 ℃,提示沙门氏菌对恶劣环境的抵抗能力强,其表面生物膜使得沙门氏菌在严酷的环境中得以生存,增加沙门氏菌到达肠道的机会,导致了食物中毒感染事件的发生[5]。
近年来,随着分子生物技术的广泛普及,实时荧光定量PCR技术已成为检测沙门氏菌的常用方法,具有敏感性高、特异性强的特点,可及时提供快速、准确的病原学诊断。本次食物中毒检测分析过程采用了18种食源性致病菌的多重PCR检测技术,可快速识别食物中毒事件感染的病原体,缩短了传统培养时间,提升了临床检测效率。实验室结果显示多重PCR结果与分离鉴定结果一致,12株沙门氏菌血清分型均为B群鼠伤寒沙门氏菌,其中7株来自7份病例肛拭子样本(含2名厨师),2株来自2份病例粪便样本,3株来自3份工作人员肛拭样本。鼠伤寒沙门氏菌作为一群泛嗜性沙门氏菌,其多重耐药性正逐年增强[6-7],在2020-2021年我国吉林省收集的133株鼠伤寒沙门氏菌药敏试验结果证实,对氨苄西林和四环素耐药的菌株比率均为73.5%,且其中多重耐药菌株高达94株,这与国内很多地区对沙门氏菌耐药性的研究类似[8]。细菌基因组所含有的耐药基因与其耐药性表型呈现良好相关性[9]。本研究发现每株鼠伤寒沙门氏菌均携带多种耐药基因,提示菌株均有形成耐药基因所针对抗生素的抗性的潜力。从耐药基因的携带推断抗菌药物的耐性来看,应用WGS预测的氨基糖苷类、头孢菌素、青霉烷、四环素的耐药性与药敏结果完全一致,与张璐等的相关研究相符[10]。同时,这9株菌株对阿米卡星、氨苄西林、头孢唑啉、庆大霉素、哌拉西林、四环素多重耐药,与全国的沙门氏菌多重耐药现象相符[11-12],提示临床应合理用药,减少耐药株的出现。此次患者治疗可首选头孢噻肟、头孢他啶、阿莫西林/克拉维酸、哌拉西林-他唑巴坦等敏感抗生素。临床选用头孢他啶静脉滴注后,患者全部治愈。
PFGE 是20 世纪80年代中期发展起来的一种非常有效的分子分型方法,被称为细菌分子生物学分型技术的“金标准”[13-14]。本文通过XbaI酶切,应用PFGE技术,对一起食物中毒分离株进行分子分型,产生的电泳条带较为理想,每株约产生12条30~700 kb条带,9株鼠伤寒沙门氏菌获得完全一致的PFGE 图谱,提示它们为同源菌株。鉴于此方法分辨能力有限,故采用了基因测序溯源的手段对菌株进行了进一步分析。分析结果显示9株沙门氏菌的MLST型别均为ST19型,其基因长度分布、类别数量分布、MLST分型管家基因序列种类编号均高度一致,为鼠伤寒沙门氏菌最常见的ST型[15]。cgMLST是使用某一个种的细菌核心基因组中的上千个基因位点的序列差异对菌株进行区分和分型,与传统的MLST分析相比具有更高的分辨力,在金黄色葡萄球菌、嗜肺军团菌、结核分枝杆菌等多种病原菌的分型和分子流行病学研究中,显示了非常好的应用前景。在本次分析的9株样本中,其cgMLST的cgST分型为264803,表明了样本之间的高度同源性。为了进一步区分样本之间的关系,我们还进一步进行了wgSNP分析。从SNP的系统发育树来看,本次食物中毒事件各菌株间SNP差异数为1~6。有文献报道,SNP差值小于21为划定暴发聚集的共同阈值[16],因此可判定本次事件为同一暴发来源,进一步明确了各菌株间关系。梁少溢等[17]研究报道,除去蝇、虫和鼠等因素外,厨师也是极易引起食物中毒事件的关键因素。2015年北京东城区咸水鸭食物中毒事件中,4名厨师均为无症状STy携带者[18]。本次事件9株测序样本中,2株来源于厨师,两者之间SNP差异个数为2,与其他样本来源菌株SNP差异数最大为6,提示厨师和该起食物中毒事件密切相关,可能是污染的来源。从进化树关系分析可以看出,9株菌自成一簇,与数据库其他沙门氏菌相差甚远,属于新的克隆分支。通过WGS序列分析发现,本次事件9株ST19型鼠伤寒沙门氏菌与江西省分离的2020年参考菌株(GCF_020221795.1)的亲缘关系接近。两者间可能存在未知途径的传播,待进一步研究考证。
本研究对鼠伤寒沙门氏菌引起的食物中毒暴发菌株从PFGE与WGS递进角度进行了分析,有助于我们迅速发现不同分离分离菌株的相关性,深度揭示细菌性传染病暴发可能来源,对食源性疫情暴发可以起到有效的控制,也可以为今后顺利开展食源性病原菌的实验室诊断和应急处置工作提供准确的依据。本次分离菌株存在多重耐药现象,提示在今后工作中应加强国家致病菌识别网监测和细菌耐药监测,指导临床有针对型的选用抗菌药物,避免多重耐药菌株的发生。本次调查存在的不足是,在应急处置过程中仅采集了肛拭子和环境样本,未进行食品样本的采集,传播证据链条还不够完整,无法判定何种途径导致病原体传播。今后,相关部门要加强对餐饮食堂的监管力度,督促其认真落实食品留样制度,合力防范食品安全事件的发生。
致谢:武汉市疾病预防控制中心病原微生物所周军波老师和费小圆老师在PFGE分型检测中给予大力帮助,在此一并致谢。
利益冲突:无
猜你喜欢
今日农业(2021年11期)2021-08-13
基层中医药(2020年5期)2020-09-11
食品与机械(2019年1期)2019-03-30
基层中医药(2018年5期)2018-08-31
兽医导刊(2016年12期)2016-05-17
制造技术与机床(2015年10期)2015-04-09
特产研究(2014年4期)2014-04-10
中国中医药现代远程教育(2014年21期)2014-03-01
云南畜牧兽医(2014年4期)2014-02-28
遗传(2014年3期)2014-02-28
