
含Cl反应力场ReaxFF的评估
2024-02-21 00:52:16肖卓君王晴晴刘艳洁
火炸药学报 2024年1期
肖卓君,肖 斌,何 祺,王晴晴,刘 馥,刘艳洁,王 宁,李 伟,刘 轶
(1.上海大学 材料基因组工程研究院,上海 200444;2.上海大学 理学院物理系,上海 200444;3.上海电子信息职业技术学院 机械与能源工程学院,上海 201411;4.湖北航天化学技术研究所,湖北 襄阳 441003)
引 言
固体推进剂作为常见的含能材料,在军事、民用领域以及土木工程等动力能源材料方面都有广泛的应用[1]。高氯酸铵(AP)是复合改性双基推进剂和复合推进剂中常用的氧化剂,被广泛运用于固体火箭推进剂、炸药和烟火药的生产中[2]。因此,理解AP热分解的微观机理对于推进剂的制备、加工、贮存、运输和使用等方面都具有重要的理论参考价值。大量实验研究[3-9]为理解AP热分解微观机理做出了探索。随着模拟手段的稳步发展,原子层次的计算方法已逐渐被用于对AP性质和分解过程进行深入研究。刘博等[10]用第一性原理方法计算AP晶体的能带结构、态密度等材料物理性能, Chu等[11]使用神经网络模型对AP分解进行模拟。
为阐明包括反应路径、中间和最终产物在内的复杂反应规律,反应分子动力学作为一种有效研究大尺度凝聚态化学反应的模拟方法,是研究含能材料热解和燃烧的有力手段。本研究通过比较力场和量子力学计算结果以及实验事实,测试了两种现有含Cl有机力场参数的有效性和适应性,并总结出了适用于模拟AP分解反应系统的力场参数优化、改进方向,为后续相关反应力场的开发奠定基础。
1 计算方法
1.1 反应力场 (Reactive Force Field)
反应分子动力学(Reactive Molecular Dynamics, RMD)方法是一类能够描述化学键光滑断裂和形成的分子模拟方法,其核心是描述原子间相互作用的反应力场(Reactive Force Field,简称ReaxFF)。ReaxFF是构建在键级(Bond Order)势函数基础上的经典分子力场,如式(1)所示,其能量表达式考虑到分子内键长(Ebond)、键角(Eval)、二面角(Etors)等对能量的贡献,以及过配位的能量矫正项(Eover)、欠配位的能量矫正项(Eunder)、键角能量的惩罚项(Epen)、共轭能量项(Econj)、分子间长程的范德华力(EvdWaals)和静电相互作用项(ECoulomb)[12]。
Esystem=Ebond+Eover+Eunder+Eval+Epen+
Etors+Econj+EvdWaals+ECoulomb
(1)
基于键级的能量泛函设计使该力场能够连续且平滑地描述化学键断裂和形成过程中的能量变化。能量泛函参数通过拟合量子力学计算数据集获得[13],在保证精度的同时获得效率上的极大提高。基于ReaxFF的MD方法无需预先假定反应机制,同时考虑温度、压力等复杂极端外界环境因素的影响。因而RMD方法是研究含能材料热解和燃烧反应的有力理论工具,该方法所能提供的信息是传统量子力学/化学反应动力学方法的有益补充。
ReaxFF在含能材料领域获得大量成功应用,包括对含能材料在机械和热冲击下化学反应的研究[14-16],以及2012年首次使用ReaxFF方法获得对自燃推进剂界面发生的物理和化学过程的原子级理解[17],至2021年对自燃双组元推进剂反应路径和机理的分析研究[18]。截至2022年底,含C/H/O/N/Cl元素的反应力场文献有19篇[19-37],其中有4篇文献对Cl元素相关参数进行了优化。
出于适用程度的考量,本研究从上述4篇优化了Cl相关参数的文献中选择了两篇文献作为参考,并在文中称之为ReaxFFC/H/O/N/Cl-2010[20]和ReaxFFC/H/O/N/Cl-2013[27]。其中ReaxFFC/H/O/N/Cl-2010力场在文献中被用于对CuCl2水溶液中的铜配合物进行表征,ReaxFFC/H/O/N/Cl-2013力场被用于模拟Cl2和HCl气体对钛金属和二氧化钛进行蚀刻。基于以上两种参考力场,使用Lammps软件对典型含Cl小分子结构进行了静态能量扫描计算,以及AP热分解过程的反应分子动力学模拟。
1.2 量子力学计算
量子力学计算中,使用数值方法求解多体薛定谔方程组可以获得体系的能量及其他性质,电子密度泛函理论(Density Functional Theory, DFT)通过构建粒子密度和体系基态物理性质的函数来简化薛定谔方程组的求解问题,是一种基于量子力学的从头算(ab-initio)理论,通常把基于密度泛函理论的计算称为第一性原理计算(first-principles)。为合理描述AP体系,本研究选择了适用于主族热化学、动力学、非共价相互作用以及价态和Rydberg态电子激发能计算的M06-2X泛函[38],以及6-311++g**基组,对含Cl的典型小分子进行了第一性原理计算。共筛选了20种含Cl典型小分子(见图1),按照元素组合类型分为Cl—H、Cl—O、Cl—C、Cl—N、Cl—Cl类以及多种元素组合类(Cl/C/H/O/N)。
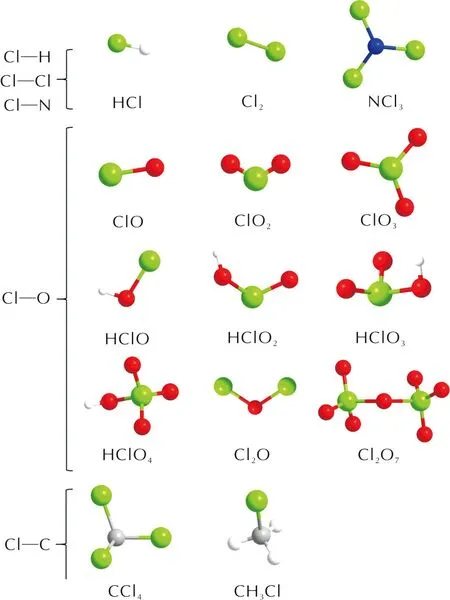
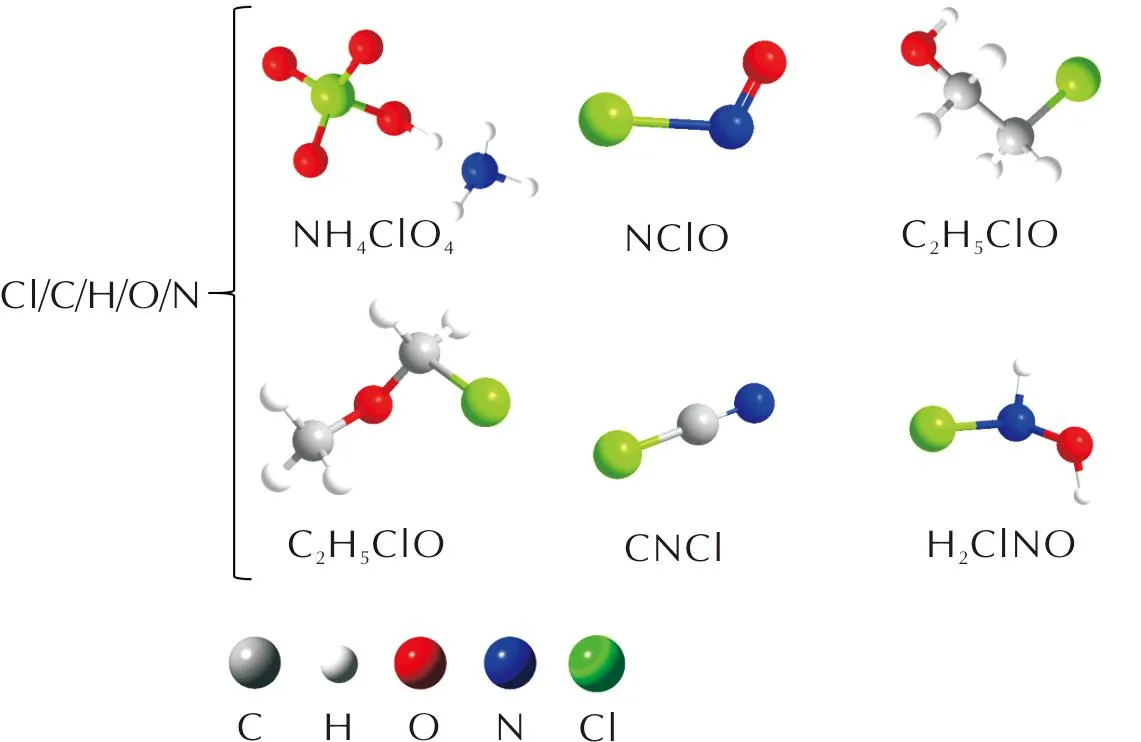
图1 20种典型含Cl小分子构型Fig.1 20 Kinds of typical configurations of small molecules containing Cl
使用Gaussian软件对每一种典型含Cl小分子进行结构优化,并计算随着分子中含Cl键长、键角、二面角大小改变时,分子能量相对于稳定结构时的变化,即能量扫描计算。能量扫描结果将作为力场计算结果的验证参考,以分析两种含Cl文献力场的适用程度。
2 结果与讨论
2.1 DFT-力场计算结果对比
针对20种典型含Cl小分子,DFT共计算了61条能量扫描曲线,总计1091个结构数据点,结果见表1。
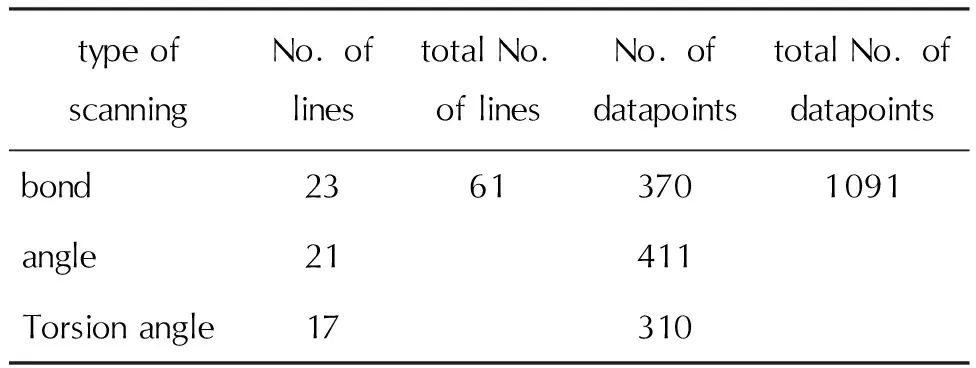
表1 不同种类的DFT能量扫描数据集统计Table 1 Statistics of different types of DFT energy scanning datasets
对于每个DFT数据点所对应的分子结构,都分别使用ReaxFFC/H/O/N/Cl-2010和ReaxFFC/H/O/N/Cl-2013力场计算其静态能量,并与DFT结果进行对比,结果见图2。
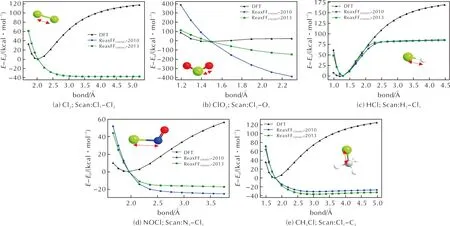
图2 键长—能量扫描曲线Fig.2 Bond length—energy scanning curves
对于Cl—H键,由图2(c)可见,在间距1.0~2.1Å内,力场ReaxFFC/H/O/N/Cl-2013的计算结果能够很好地与DFT计算结果吻合,而力场ReaxFFC/H/O/N/Cl-2010吻合欠佳。
对于Cl—Cl、Cl—C,如图2(a)、(e)所示,两种力场并未出现明显势阱,且随着原子间距的增加,DFT与力场计算结果偏差更加明显。类似地,两种文献力场在计算Cl与O/N两种原子键距对能量的影响时,也与DFT计算结果有较大偏差,见图2(b)和(d)。以图2(b)中ClO2分子能量随Cl-O键长变化曲线为例,一个影响较大的因素在于:两种力场均无法正确描述能量随键长变化的定性关系,即力场计算结果中能量随键长呈单一的负相关变化趋势,没有平衡位置的出现,而这显然与常规势能曲线不符。该误差在后续的分子动力学模拟中则体现为:为了趋向更低的势能值,Cl原子与O原子距离过大而无法正常成键,从而影响了含Cl—O键的相关中间体与产物的形成。这一点可以从RMD的物种分析结果获得验证:两种力场模拟的反应过程中均无ClO2生成。
在研究键角对能量的影响时, ReaxFF反应力场也表现出与DFT方法差异较大的计算结果,大部分键角—能量曲线如图3(a)所示,力场无法合理描述能量随键角的变化趋势。少数情况下如图3(b)、(c)、(d)所示,在ReaxFFC/HO/N/Cl-2010力场仍然无法正确地描述能量随键角变化的定性关系的情况下,ReaxFFC/HO/N/Cl-2013力场计算结果与DFT结果更加接近。
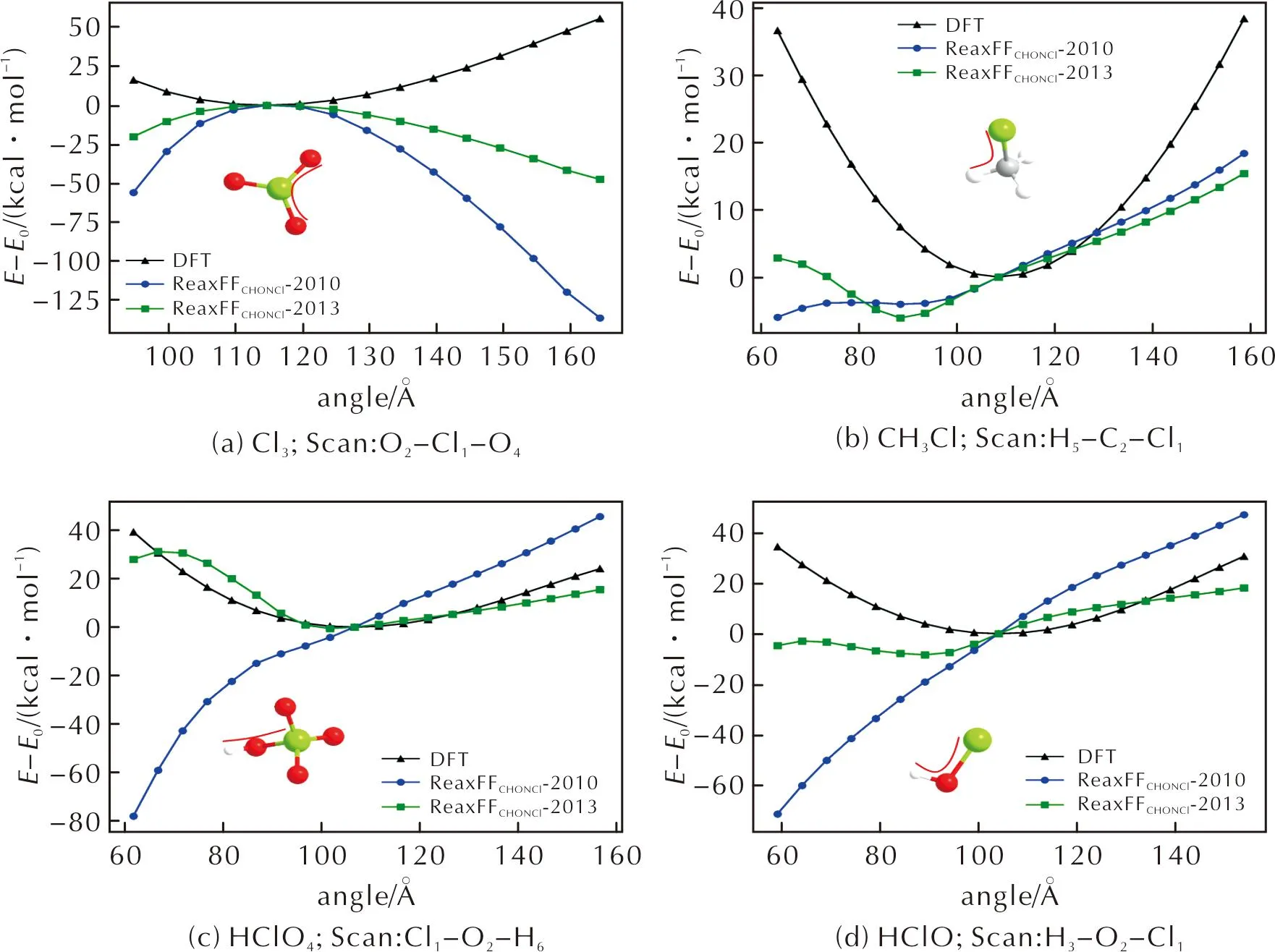
图3 键角—能量扫描曲线Fig.3 Bond angle—energy scanning curves
二面角-能量扫描曲线(见图4)与键角-能量变化曲线类似,大多数结果中,两种力场均无法定性描述曲线趋势,如图4(a)。但值得一提的是,如图4(b)、(c)所示,对于分子C2H5ClO和CH3Cl中的二面角C—Cl—C—H,两种力场的计算结果都与DFT值吻合较好。
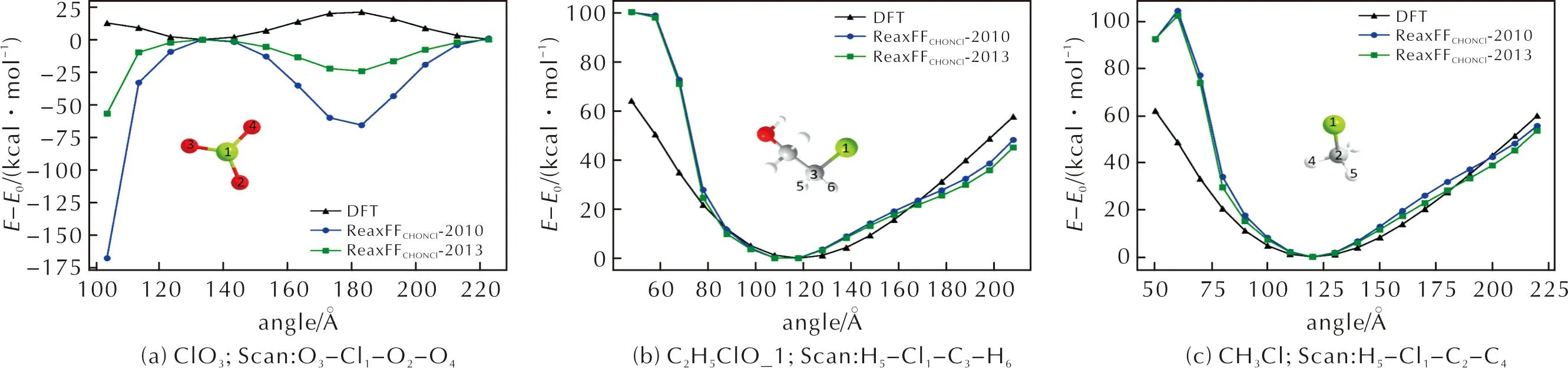
图4 二面角—能量扫描曲线Fig.4 Torsion angle—energy scanning curves
结合图5(d)和(e)小分子的ADCH电荷计算结果,从以上对比中还可发现,与DFT值吻合较好的力场计算结果,均是含低价态Cl小分子的扫描曲线。其中包括HCl分子的H—Cl键长扫描、C2H5ClO和CH3Cl分子的C—Cl—C—H二面角扫描。相反地,以图5(a)、(b)、(c)中AP分子的各类能量扫描为例,含高价Cl的小分子,包括AP、ClO2等分子的各项扫描结果,与DFT计算结果对比均出现了不同程度的误差。
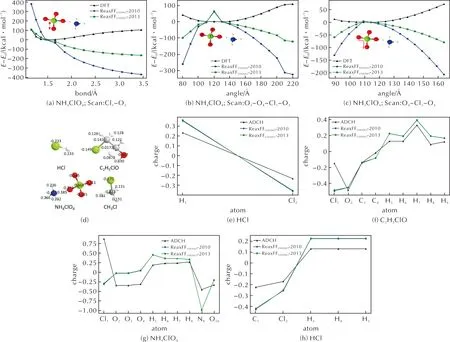
图5 AP小分子中:(a)键长-能量扫描曲线;(b)二面角-能量扫描曲线;(c)键角-能量扫描曲线;(d)HCl/C2H5ClO/NH4ClO4/CH3Cl中各原子的ADCH电荷值;(e)HCl/C2H5ClO/NH4ClO4/CH3Cl中各原子ADCH电荷与ReaxFF力场计算电荷值Fig.5 In AP small molecules, (a) Bond length -Energy scanning curves; (b) Torsion angle-energy scanning curves; (c) Bond angle-energy scanning curve; (d) ADCH charge values of each atom in HCl/C2H5ClO/NH4ClO4/CH3Cl; (e) ADCH charge values and the results from ReaxFF of each atom in HCl/C2H5ClO/NH4ClO4/CH3Cl
2.2 反应分子动力学模拟
实验结果分析认为,AP在常温常压下以晶体形式存在,但在分解初期存在离解与升华现象[3],本研究的分子动力学模拟旨在探索力场的适用程度,分析力场是否能对相关产物的形成进行合理描述,故采用无序随机的小分子排布方式进行建模,如图6所示。
图6 (a)RMD中AP小分子无序排布模型;(b)AP分子Fig.6 (a) Model of disordered arrangement of AP small molecules in RMD (b) AP molecule
模拟中总计400个AP小分子,取实验密度1.95g/mL作为初始密度,使用ReaxFFC/H/O/N/Cl-2010和ReaxFFC/H/O/N/Cl-2013力场参数,积分步长0.1fs,NVT系综下分别加热300ps至1500,2000和2500K,并分别于目标温度下NVT保温500ps,分析不同温度下两种反应力场模拟AP分解时生成的产物。
早期对AP分解产物的研究发现,热分解的产物成分取决于温度[3, 7, 9, 39-40],分为低温和高温两个阶段,两个阶段的分解机理不同。基于本研究模拟AP分解的升温范围,认为反应属于高温阶段。在温度高于380℃时,AP分解反应方程式可参考:

此外,高温阶段还检测到了NH3、ClO2、HCl、NOCl、N2等多种产物。
O2作为AP高温分解阶段的主要生成物之一[8],在两种力场模拟的产物中均有体现,如图7(a)和(b)所示。同样地,两种力场模拟结果均显示了H2O的生成,如图7(c)和(d)所示,与实验结果一致。
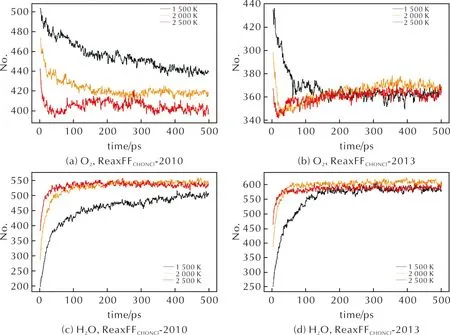
图7 ReaxFFC/H/O/N/Cl-2010力场MD模拟中,(a)O2、(c)H2O分子数量随时间的变化曲线;ReaxFFC/H/O/N/Cl-2013力场MD模拟中,(b)O2、(d)H2O分子数量随时间的变化曲线 Fig.7 In MD with ReaxFFC/H/O/N/Cl-2010, curves of(a) O2, (c) H2O as a function of time. In MD with ReaxFFC/H/O/N/Cl-2013, curves of (b) O2, (d) H2O as a function of time
根据文献[41]由实验结果推测的AP高温反应过程,温度对产物中O2的含量并无明显影响,ReaxFFC/H/O/N/Cl-2013力场的模拟结果更符合这一推论。

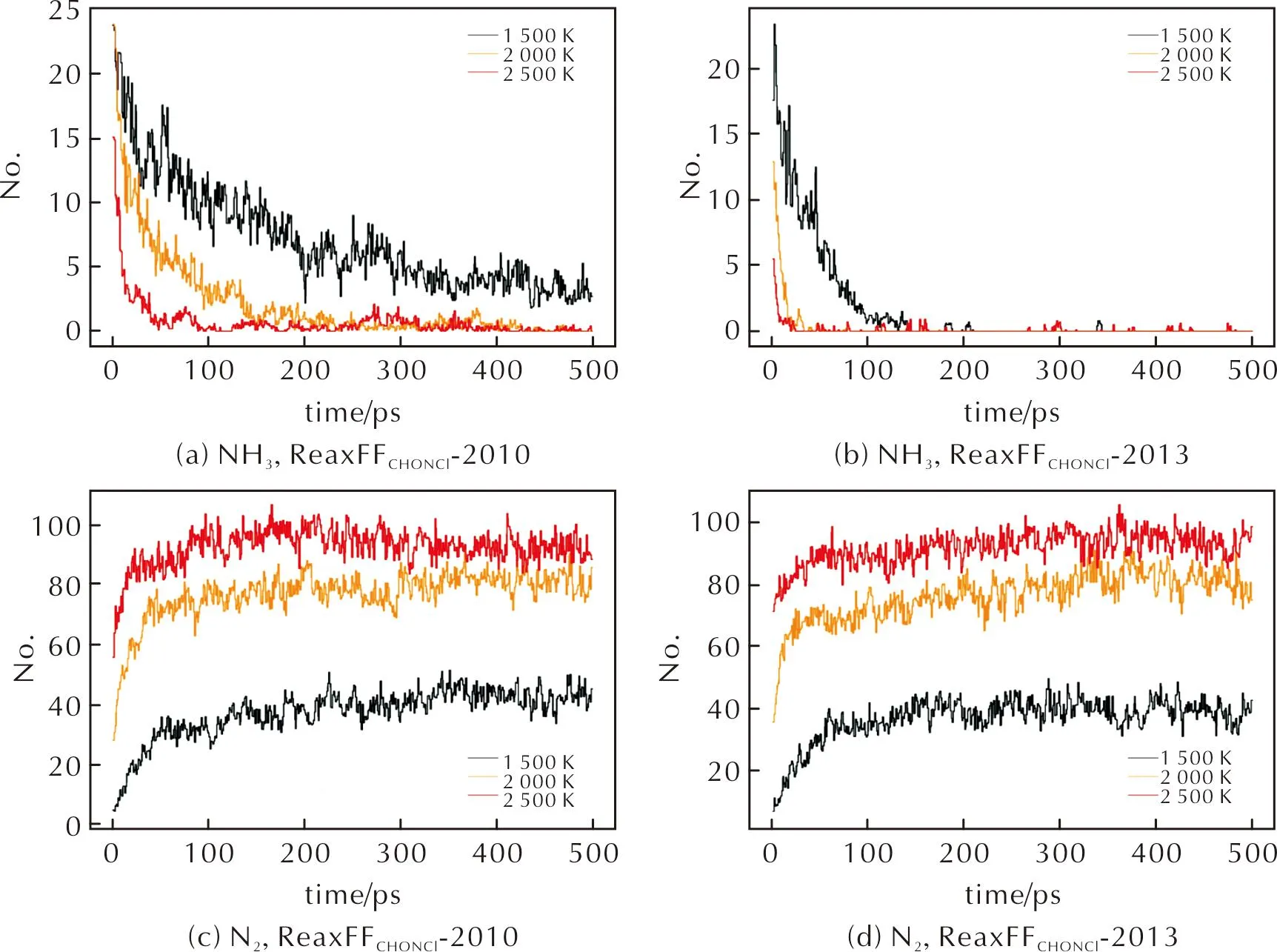
图8 ReaxFFC/H/O/N/Cl-2010力场和ReaxFFC/H/O/N/Cl-2013力场MD模拟中NH3和N2分子数量随时间的变化曲线Fig.8 In MD with ReaxFFC/H/O/N/Cl-2010 and ReaxFFC/H/O/N/Cl-2013, curves of NH3 and N2 as a function of time
图9为N2O和NO2中键长—能量扫描曲线的DFT和力场计算结果对比。在力场模拟产物中,大量N原子以N2的形式出现,如图8(c)、(d)所示,而AP高温下快速热分解实验中[9],N原子最终以NO2、NOCl、N2O以及少量NO的形式出现在产物中,其中NO2、N2O、NOCl这三类分子均未在力场模拟结果中体现。从图9可以推测,与Cl—O键类似,力场对N—O成键行为的描述存在不足,导致N、O原子在分子动力学模拟过程中难以成键。
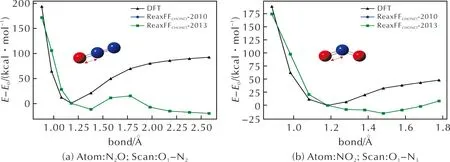
图9 N2O、NO2中N—O键长—能量扫描曲线Fig.9 Bond-Energy scanning curve of N—O in N2O, NO2
图10分别为两种力场模拟中,HCl和Cl数量随时间的变化曲线。
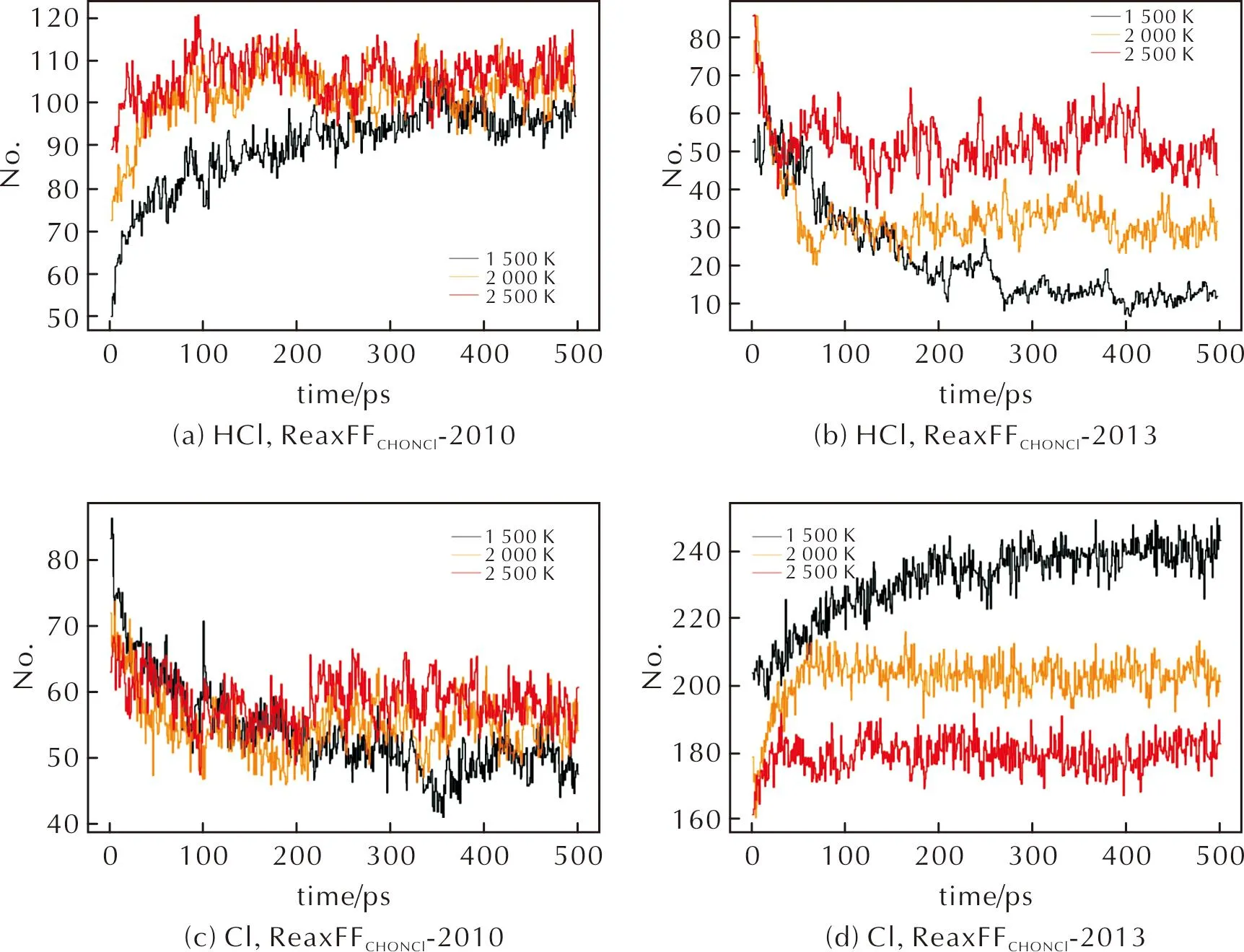
图10 ReaxFFC/H/O/N/Cl-2010力场和ReaxFFC/H/O/N/Cl-2013力场模拟中HCl和Cl数量随时间的变化曲线Fig.10 In MD with ReaxFFC/H/O/N/Cl-2010 and ReaxFFC/H/O/N/Cl-2013, curves of HCl and Cl as a function of time
对于含Cl分解产物,两种力场均模拟出HCl分子的生成,且HCl的含量随着温度升高而增大,见图10(a)和(b),这一点与高温阶段实验事实相符。然而,实验显示[43],高温情况下,Cl原子主要以Cl2的形式释放,HCl分子作为产物并非主要类型。但在RMD模拟中,Cl原子主要以HCl的形式出现在产物里。这一模拟结果很大程度上源于力场文件中H—Cl参数能够较为准确地描述H原子和Cl原子的成键情况,而Cl—Cl描述欠佳,从图2(a)Cl—Cl键长的能量扫描结果对比图也能够看出,两种参考力场下Cl原子均倾向于远离而非成键。
除HCl分子,其他大多数Cl原子还以游离的单个原子状态而非Cl2单质形式出现在力场模拟产物中,见图10(c)、(d),这与力场中Cl—Cl键长参数的局限性有很大关联。其次,在AP分解过程中,典型的含高价Cl中间体并未在力场计算结果中观察到,包括ClO2、ClO3、HClO4等。说明现有的Cl元素相关参数对原子间作用力的描述仍不够准确,还存在较大可供优化的空间。
AP具有明显区别于其他推进剂含能材料的性质。首先,AP分子中Cl元素的化合价为+7价,具有极强的氧化性。大多数含能材料仅由C/H/O/N元素组成,高价态Cl的存在使得AP与其他含能材料在成分上有显著差异。其次,AP分子包含H/O/N/Cl四种元素,如果考虑这些元素所有可能存在的氧化态,则AP的分解可能存在很多的过程。多种因素影响使得对AP分解过程进行分子动力学模拟时,从其他研究中迁移而来的力场可能存在无法合理描述在快速热解反应过程中高价态Cl元素与其他有机元素的耦合反应行为问题。同时,专门针对含能材料的可用含Cl力场目前仍未被开发出来[44]。为指导开发含Cl有机反应力场,本研究使用两种参考力场,计算了典型含Cl小分子的能量扫描曲线,以及AP分解的RMD模拟,并将力场计算结果分别与DFT结果、实验结果进行对比评估。
分析比较能量扫描曲线和DFT量子力学计算结果,发现 ReaxFFC/H/O/N/Cl-2013力场的H—Cl参数表现更好,对含H、Cl的小分子(如HCl、CH3Cl)的成键断键情况,以及部分二面角对能量的影响有较为合理的预测能力。然而,对于含高价Cl小分子,两种力场均无法恰当地描述原子间距以及价角、二面角对能量的贡献。其次,从分子动力学模拟分解产物和实验观测结果的对比分析得出,现有的含Cl有机力场对AP分解反应系统中非含Cl小分子(例如H2O、N2、NH3等)的行为有一定的复现能力,其中ReaxFFC/H/O/N/Cl-2013力场对HCl、部分非含Cl分解产物的模拟表现更加贴合实验事实,反之,对于含高价Cl的小分子产物,以及含N/O元素的产物,由于力场对Cl—C/O/N原子之间、N—O原子之间缺乏适度的成键描述,使得分解反应模拟中难以形成含高价Cl小分子、NO2、N2O等产物,导致对实验结果重要部分的复刻缺失。
此外,在两种参考力场的分子动力学模拟过程中均观察到,AP小分子群在加热过程中就开始了反应,推测原因在于两种力场的参数均未添加色散修正项(即—lg修正项[45]),导致加热过程中NVT压强过高,促使反应的发生。
3 结 论
(1)基于量子力学密度泛函理论计算,建立了含Cl有机小分子的数据集,并对现有文献中的反应力场的表现进行了评估研究。
(2)本研究的评估结果对于使用ReaxFF反应力场模拟AP分解提供了重要警示:目前文献中的ReaxFF力场虽然含有C/H/O/N/Cl参数,但对于较为特殊的AP体系,本工作证明了Cl参数的迁移性并不理想,与密度泛函计算结果相比误差较大。主要误差来源于能量表达式中分子内键长、键角和二面角等因素。因此,优化现有力场参数,特别是键长、键角和二面角的能量项相关参数,发展适用于高价Cl元素和氮氧化物NOx体系的力场参数,成为使用ReaxFF反应力场模拟AP复杂分解反应的迫切需求。本工作为未来开发更准确的含Cl反应力场提供了前期基础和建议方向。
猜你喜欢
中学化学(2024年3期)2024-06-30 15:19:19
航空材料学报(2023年6期)2023-12-18 05:23:50
小学生学习指导(小军迷联盟)(2023年3期)2023-03-27 09:22:44
高中数理化(2022年16期)2022-09-14 13:57:06
中国音乐学(2022年1期)2022-05-05 06:48:46
中学生数理化(高中版.高考数学)(2022年2期)2022-04-26 14:05:00
新世纪智能(数学备考)(2021年4期)2021-08-06 09:04:40
中学生数理化(高中版.高考数学)(2020年3期)2020-05-25 06:53:20
青岛大学学报(工程技术版)(2019年2期)2019-09-10 07:22:44
中学生数理化(高中版.高考理化)(2019年6期)2019-06-22 09:55:38
