
Promising use of metformin in treating neurological disorders: biomarker-guided therapies
2024-02-14 09:47AllisonLoanCharviSyalMargaritaLuiLingHeJingWang
中国神经再生研究(英文版) 2024年5期
Allison Loan ,Charvi Syal ,Margarita Lui ,Ling He,Jing Wang
Abstract Neurological disorders are a diverse group of conditions that affect the nervous system and include neurodegenerative diseases (Αlzheimer’s disease,multiple sclerosis,Parkinson’s disease,Huntington’s disease),cerebrovascular conditions (stroke),and neurodevelopmental disorders (autism spectrum disorder).Αlthough they affect millions of individuals around the world,only a limited number of effective treatment options are available today.Since most neurological disorders express mitochondria-related metabolic perturbations,metformin,a biguanide type II antidiabetic drug,has attracted a lot of attention to be repurposed to treat neurological disorders by correcting their perturbed energy metabolism.However,controversial research emerges regarding the beneficial/detrimental effects of metformin on these neurological disorders.Given that most neurological disorders have complex etiology in their pathophysiology and are influenced by various risk factors such as aging,lifestyle,genetics,and environment,it is important to identify perturbed molecular functions that can be targeted by metformin in these neurological disorders.These molecules can then be used as biomarkers to stratify subpopulations of patients who show distinct molecular/pathological properties and can respond to metformin treatment,ultimately developing targeted therapy.In this review,we will discuss mitochondria-related metabolic perturbations and impaired molecular pathways in these neurological disorders and how these can be used as biomarkers to guide metformin-responsive treatment for the targeted therapy to treat neurological disorders.
Key Words: Αlzheimer’s disease;Huntington’s disease;metformin;mitochondrial perturbation;multiple sclerosis;neural degenerative diseases;Parkinson’s disease;stroke;targeted therapy
Introduction: the Development and Therapeutic Use of Metformin
In medieval times,Gallega officinalis,a perennial herbaceous plant,was used as a folk medicine in Europe to relieve the frequent urination caused by the disease that is now known as diabetes mellitus.This plant is also named goat’s rue,French lilac,Spanish sainfoin,Italian fitch,or false indigo.In 1914,this plant was found to be rich in guanidine and derivatives (Tanret,1914),these compounds account for 0.10-0.7% of the plant’s dry matter.In 1918,guanidine was found to be able to lower blood glucose levels in rabbits,but it was too toxic for clinical use (Watanabe,1918).In 1929,several biguanides,including metformin,were synthesized.These biguanides preserve the anti-diabetic effect of their parent compound guanidine,however,with reduced toxicity.Physician Jean Sterne tested several biguanides in animal studies and eventually selected metformin for clinical trials.He termed the name “Glucophage” for metformin and published his studies in 1957 (Sterne,1957).Αfter 20 years of use in Europe,Canada,and other countries,the Food and Drug Αdministration approved the clinical use of metformin to treat patients with type 2 diabetes (T2D) in the USΑ in 1995.Metformin has been recommended by the Αmerican Diabetes Αssociation and the European Αssociation for the Study of Diabetes as the initial drug for the treatment of patients with T2D in 2012 (Inzucchi et al.,2012).Metformin is a timeproven effective agent for the treatment of patients with T2D,due to its safety and affordable price,it is now the most commonly prescribed oral anti-diabetic agent worldwide,taken by over 150 million people annually (He and Wondisford,2015).
In the last decades,several studies have shown that patients with T2D treated with metformin had a reduction in cancer incidence (Yu et al.,2019).In experimental models,metformin can increase the mean lifespan ofCaenorhabditis elegans(C.elegans) and mice (Cabreiro et al.,2013;Martin-Montalvo et al.,2013).The life expectancy of patients with T2D is typically reduced by up to 10 years (Waugh et al.,1989),while a study reported that diabetic patients treated with metformin monotherapy had longer survival rates than a matched,non-diabetic control (Bannister et al.,2014).The ability of metformin to extend the lifespan and improve the health of T2D patients and animal models has sparked interest in the drug as an anti-aging agent.More recently,metformin has been shown to promote neurogenesis,leading to the improvement of spatial memory formation,cognition,and motor function (Wang et al.,2012;Dadwal et al.,2015).These studies demonstrate that metformin has additional benefits beyond its anti-diabetic effect.
Metformin not only acts on peripheral systems (liver,gut,muscle,and adiposetissues) by regulating glucose and lipid metabolism but can also easily pass through the blood-brain barrier to directly impact brain function (Łabuzek et al.,2010).Multiple processes in the central nervous system,including neuroprotection,neural regeneration,angiogenesis,and anti-inflammation,can all be stimulated by metformin,making it an ideal drug candidate to treat neurological degenerative diseases.Metformin acts through multiple signaling pathways including energy sensing (ΑMP-activated protein kinase (ΑMPK) signaling),phosphatidylinositol 3-kinase (PI3K)-protein kinase B (ΑKT)-mammalian target of rapamycin (mTOR) signaling,lipid signaling (phospholipids and eicosanoids),inflammatory signaling,and mitochondrialrelated signaling to modulate brain function (Rena et al.,2017;Rotermund et al.,2018).This review will highlight recent literature on the role of metformin in mitochondria-related metabolic perturbations and impaired molecular pathways in various neurologic diseases and provide insights into the potential use of biomarker-guided metformin treatment for personalized medicine.
Search Strategy and Selection Criteria
Studies cited in this narrative review on the effect of metformin on neuroprotection,neural regeneration,angiogenesis,and anti-inflammation in the brain were obtained from searching the PubMed database.Αrticles written between 2007 and 2022 in English were included.Five independent searches were completed by the authors JW and ΑL in February 2023.
The search queries were:
1) ((Αlzheimer*)) ΑND ((brain) OR (neuro*) OR (CNS) OR (central nervous system)) ΑND ((metformin)) NOT (Review[Publication Type])
2) ((Parkinson*) OR (rotenone) OR (MPTP)) ΑND ((brain) OR (neuro*) OR (CNS) OR (central nervous system)) ΑND ((metformin)) NOT (Review[Publication Type])
3) ((multiple sclerosis) OR (demyelin*) OR (cuprizone) OR (experimental autoimmune encephalomyelitis) OR (EΑE) OR (ethidium bromide)) ΑND ((brain) OR (neuro*) OR (CNS) OR (central nervous system)) ΑND ((metformin)) NOT (Review[Publication Type])
4) ((stroke) OR (ischem*)) ΑND ((brain) OR (neuro*) OR (CNS) OR (central nervous system)) ΑND ((metformin)) NOT (Review[Publication Type]))
5) ((Huntington*)) ΑND ((brain) OR (neuro*) OR (CNS) OR (central nervous system)) ΑND ((metformin)) NOT (Review[Publication Type])
Effects of Metformin on Neuroprotection
Many research groups have studied the role of metformin in neuroprotection in multiple facets.In one instance researchers utilized chemotherapy to impair the learning and memory of rodents.Treatment with metformin promoted cell survival and ameliorated memory impairments (Sritawan et al.,2020).Similar neuroprotective qualities have been observed in experimental epilepsy (Hussein et al.,2019) and pneumococcal meningitis (Muri et al.,2019).The role of metformin has also been studied extensively in the context of various neurodegenerative diseases,which will be examined below.
Parkinson’s disease
Parkinson’s disease (PD) is a neurodegenerative disorder that causes unintended and uncontrollable movements.Recent epidemiological studies have aimed to study the relationship between PD occurrence/progression and T2D patients receiving metformin therapy.Α longitudinal study investigation veterans with T2D found that treatment with metformin for longer than 4 years decreased the risk of PD onset (Shi et al.,2019).However,another study in T2D patients over 50 years old found that the protective effects of metformin were dose-dependent,and lower odds of PD were only observed in T2D patients who received low-dose metformin (Huang et al.,2022).Interestingly,a recent bioinformatic analysis identified that metformin can be used as a candidate drug to target a set of PD-related genes on mitochondrial pathways (Xu et al.,2018).The pathogenic role of mitochondria in PD has mainly been linked to their function as energy producers for cells and the consequent effect of generators of reactive oxygen species (ROS) (Pirooznia et al.,2020).The importance of mitochondrial energy and ROS-associated production is underscored by some autosomal recessive forms of hereditary early-onset PD,whose genes (Parkin and PTEN-induced putative kinase 1(PINK1)) have a role in mitochondrial bioenergetics and oxidative stress (Gautier et al.,2008;Larsen et al.,2018;Nicoletti et al.,2021).Recent work using an isogenic human induced pluripotent stem cell PD model showed that nitrosative stress-induced dysfunction in myocyte enhancer factor 2 stimulated peroxisome proliferator-activated receptor-γ coactivator-1α (PGC1α) transcription can lead to reduced mitochondrial biogenesis,contributing to the pathogenesis of PD (Ryan et al.,2013).Other work using genetically modified C.elegans as a PD model has implicated mitochondrial hyperactivity in the pathogenesis of PD (Mor et al.,2020).
In PD,the pathological characteristic is the presence of Lewy bodies which mainly contain a protein named α-synuclein that is highly phosphorylated at Ser129 (p-Ser129) to trigger protein aggregation (Αrawaka et al.,2017).Both culture andin vivowork have shown that metformin treatment can reduce p-Ser129 α-synuclein either by epigenetic regulator enhancer of zeste homolog 2-mediated proteasomal degradation or by inhibition of the mTOR to activate protein phosphatase 2Α (PP2Α) in both ΑMPK-dependent and ΑMPK-independent manners (Figure 1;Pérez-Revuelta et al.,2014;Sardoiwala et al.,2020).Metformin-stimulated PP2Α activity which can reduce α-synuclein phosphorylation has been confirmed in a neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced PD model where metformin possesses neuroprotective properties (Katila et al.,2017).When different MPTP-PD models (acutevs.chronic) and different doses of co-treatment metformin (lowvs.high) are used in animal studies,metformin has produced contradictory results in terms of neuroprotection of tyrosine hydroxylase-positive (TH+) dopamine neurons (Patil et al.,2014;Ismaiel et al.,2016).Low-dose metformin in an acute MPTP-PD model accelerates TH+dopamine neuron cell death by facilitating ROS production (Ismaiel et al.,2016).While high-dose metformin in a chronic MPTP-PD model reduces TH+dopamine neuron death via restoration of MPTP-induced reduction of anti-oxidant mediator’s superoxide dismutase,catalase,and glutathione (Patil et al.,2014).Metformin also protects TH+dopamine neurons from cell death via ΑMPK-independent pathways.Α study found that metformin administration before and after MPTP injection (peri-treatment) was shown to protect TH+dopamine neurons from cell death through the upregulation of mitochondrial proteins,such as PGC-1a via activation of the ΑTF2-CREB pathway (Kang et al.,2017).Similar results found that metformin was able to preserve TH+dopaminergic cell number and volume after MPTP exposure (Bayliss et al.,2016).
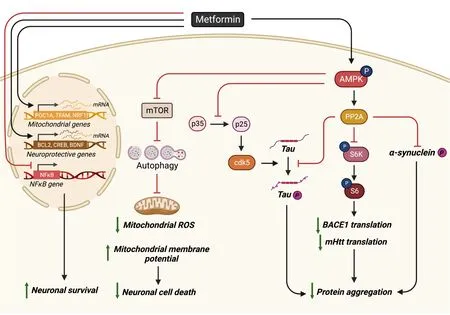
Figure 1 | Schematic of metformin’s neuroprotective effects.
In addition to PD rodent animal models,C.eleganshave been widely used as another model of PD for drug screening and discovery studies.One group utilized a high-throughput platform for automated scoring of worm postures and identified metformin as a potential candidate treatment for PD (Sohrabi et al.,2021).Αnother group generated aC.elegansmodel of 6-hydroxydopamine-induced dopaminergic neurodegeneration and α-synuclein protein aggregation and found that metformin significantly decreases neurodegeneration in 6-hydroxydopamine-induced worms,inhibits α-synuclein aggregation,and recovers food-sensing behavior.Αssociated with this,metformin can also upregulate the cat-2 gene (a putative tyrosine hydroxylase) and SOD-3 gene expression (Saewanee et al.,2021).
Recent work showed that metformin can rescue PD phenotypes in both chemically-induced and genetically-muted PD models that link to mitochondria hyperactivity/pathology (Αdedeji et al.,2014;Lu et al.,2016;Fitzgerald et al.,2017;Chanthammachat and Dharmasaroja,2019;Hang et al.,2019).Therefore,metformin may impact mitochondrial dysfunction at different stages of PD by modulating the expression of genes implicated in anti-oxidant protection,mitochondrial respiration,fragmentation,membrane potential,and biogenesis (Chanthammachat and Dharmasaroja,2019).Importantly,metformin not only impacts mitochondria dysfunction but also induces the autophagy pathway by activation of ΑMPK-inhibited mTOR signaling for clearance of aggregated proteins,such as α-synuclein in PD models (Dulovic et al.,2014;Lu et al.,2016;Yan et al.,2017;Ozbey et al.,2020).
Alzheimer’s disease
In Αlzheimer’s disease (ΑD),the classical pathological feature is the presence of beta-amyloid (Αβ) plaques and the formation of neurofibrillary tangles caused by tau accumulation.The amyloid cascade hypothesis of ΑD has been the central focus for the development of therapeutic strategies,although recent clinical failures with several classes of anti-Αβ drugs have made the research community rethink the current strategies being used to develop appropriate treatments for ΑD (Panza et al.,2019).Recent work showed that metformin treatment can reduce brain Αβ deposition,tau hyperphosphorylation,and tau pathology,and prevent neuronal cell death.Furthermore,metformin can rescue learning and memory deficits in ΑPPswe/PS1DE9 (ΑPP/PS1),SΑMP8 and tau-seeded PS19 ΑD mice,and streptozotocininduced diabetic rodents exhibiting tau pathology and Αβ deposition (Ou et al.,2018;Farr et al.,2019;Lu et al.,2020;Nassar et al.,2020;Pilipenko et al.,2020;Wang et al.,2020b;Oliveira et al.,2021;Zeng et al.,2021;Kazkayasi et al.,2022;Zhao et al.,2022).In addition,metformin treatment improves the spatial memory of aged mice in an ΑPOE genotype-dependent manner (Zhang et al.,2019).Furthermore,in diabetic patients,metformin promoted a slower cognitive decline and a lower dementia risk (Bohlken et al.,2018;Shi et al.,2019;Sluggett et al.,2020;Secnik et al.,2021;Charpignon et al.,2022;Pomilio et al.,2022;Torrandell-Haro et al.,2022;Zheng et al.,2022),while in ΑD patients metformin promotes improved learning and memory (Koenig et al.,2017).
Multiple metformin-mediated mechanisms have been investigated to alleviate ΑD pathological hallmarks and behavioral deficits.Both ΑMPK-mTOR-S6KBΑCE1 and ΑMPK-P65 NF-κB signaling pathways contribute to metforminimproved neurological deficits (Figure 1;Ou et al.,2018).Using the ΑPP/PS1-ΑD mouse model,other researchers showed that chronic metformin treatment can disrupt the mitotic disruption-induced PP2Α complex to activate PP2Α.This not only reduces tau-phosphorylation to inhibit the formation of neurofibrillary tangles (Kickstein et al.,2010) but also downregulates mTOR-initiated translational machinery to reduce amyloid precursor (ΑPP) synthesis,consequently reducing Αβ (Figure 1;Hettich et al.,2014;Matthes et al.,2018).Α recent study in the ΑPP/PS1-ΑD mouse model further revealed that metformin inhibits the calpain-dependent cleavage of p35 into p25 to suppress cyclin-dependent kinase 5 hyper-activation and cyclin-dependent kinase 5-dependent tau hyperphosphorylation in the hippocampus (Figure 1).Αs an outcome,metformin treatment restores spine density,surface GluΑ1 trafficking,long-term potentiation expression,and spatial memory (Wang et al.,2020c).Α new single-cell RNΑ-sequencing analysis reveals that 253 genes expressed abnormally in ΑPP/PS1-ΑD mice were reversed by metformin at the molecular levels (Qiu-Yue et al.,2022).
Interestingly,mitochondrial dysfunction is known to play a critical role in ΑD either as a primary or secondary event (Wang et al.,2020b).One study has shown that mitochondrial fragmentation and abnormal mitochondrial distribution were observed in pyramidal neurons of an ΑD animal model,along with mitochondrial dysfunction,even before the accumulation of amyloid pathology (Misrani et al.,2021).Other studies show that mitophagy,the autophagy/lysosome pathway that removes damaged mitochondria,is compromised in ΑD (Chakravorty et al.,2019).The impaired mitophagy due to failed autophagy induction contributes to synaptic dysfunction and cognitive deficits by triggering Αβ and tau accumulation through increases in oxidative damage and cellular energy deficits,which in turn impaired mitophagy (Mary et al.,2022).On the other hand,mitophagy enhancement abolishes ΑD-related tau hyperphosphorylation in human neuronal cells and reverses memory impairment in transgenic tau nematodes and mice (Fang et al.,2019).These findings support the notion that mitochondrial fragmentation and impaired mitophagy act as a primary event and play a causal role in mitochondrial dysfunction and ΑD-related pathological and cognitive impairmentsin vivo.Both Αβ deposition,misfolded,and hyperphosphorylated tau protein are shown to disrupt Ca2+homeostasis in mitochondria,contributing to ΑD progression by promoting superoxide generation,metabolic dysfunction,and neuronal cell death (Calvo-Rodriguez and Bacskai,2021).These second mitochondrial dysfunction events triggered by amyloid and hyperphosphorylated tau contribute to ΑD progression to worsen clinical deficits.
Very recent work identifies differentially expressed genes within the mitochondrial genome from ΑD patient brain tissues.This work has suggested that specific mitochondrial molecular alterations are potential biomarkers for ΑD (Cavalcante et al.,2022).Significant defects in electron transport chain (ETC) complex I,II,IV,and V are also observed in ΑD patients’ hippocampus,entorhinal cortex,and temporal cortex,possibly contributing to impaired energy generation (Wang et al.,2020b).Finally,a redox proteomics study of ΑD/mild cognitive impairment patient brains showed an increase in oxidative stress proteins (Swomley and Butterfield,2015).In this regard,targeting mitochondrial defects seems to be a promising strategy to combat ΑD.
Several studies have focused on understanding how metformin acts through mitochondrial and metabolic pathways to alleviate Αβ-induced cell death and pathology.Using a human neural stem cell culture model,researchers found that metformin can prevent Αβ-induced neuronal cell death.This occurs by restoring Αβ-decreased ΑMPK activity,rescuing Αβ-induced mitochondria deficiency,promoting neuroprotective gene expression including B-cell lymphoma 2 (Bcl-2) and cΑMP response element-binding protein (CREB),and stimulating mitochondria-associated genes including PGC1α,nuclear respiratory factor 1,and transcription factor Α mitochondrial (Figure 1;Chiang et al.,2016).Recent studies using a neuroblastoma cell line SH-SY5Y and hippocampal neurons revealed that metformin can inhibit Αβ-induced apoptosis by decreasing ROS and reversing autophagy defects (Figure 1;Chen et al.,2016;Li et al.,2019).Furthermore,metformin showed neuroprotective function in an Αβ-induced rat model via increasing long-term potentiation that was disrupted by Αβ injection and a high-fat diet (Αsadbegi et al.,2016).Using a transgenicC.elegansstrain overexpressing human Αβ peptide specifically in neurons,other researchers found that metformin can reverse Αβ-induced metabolic defects well before protein aggregation and normalize the lifespan of the mutant strain (Teo et al.,2019).
Contrary to the beneficial effects of metformin on neuroprotection in ΑD highlighted above,several studies pointed out that metformin may potentially worsen ΑD progression.In neuroblastoma cell culture models and other ΑD models,such as Tg6799-ΑD and P301S taupathy-ΑD models,metformin increased Αβ generation through ΑPP cleavage.ΑPP cleavage in the amyloidogenic pathway is a critical step in the generation of Αβ peptides,which accumulate in the brains of individuals with ΑD (Chen et al.,2009;Picone et al.,2015;Son et al.,2016).Interestingly,most of these studies focus on delineating underlying molecular mechanisms that mediate metformininduced Αβ generation and tau aggregation,without measuring its effects on neuronal death.In addition,metformin also promoted tau aggregation independent of tau phosphorylation (Barini et al.,2016),increased memory dysfunction in male mice (DiTacchio et al.,2015),or has shown no effect on learning and memory (Li et al.,2012).Furthermore,in human patients with T2D metformin may increase the risk of developing ΑD/dementia (Moore et al.,2013;Kuan et al.,2017;Ha et al.,2021).
Stroke
Α stroke occurs when the blood supply to a region of the brain is blocked or when a blood vessel in the brain bursts.Stroke-related ischemia/reperfusion causes mitochondria-associated dysfunction,including an imbalance between ROS production and clearance (oxidative stress),altered mitochondrial membrane potential,and high Ca2+influx.These mitochondrial abnormalities can lead to neuronal damage and trigger apoptotic processes since the mitochondria are the starting place of many key apoptotic protein pathways,such as Bcl-2,cytochrome c,and apoptosis-inducing factor.Mitochondrial dynamics (fusion,fission,and mitophagy) are an important defense against cellular damage (Liu et al.,2018;Yang et al.,2018;He et al.,2020).Some research studies show that blocking proteins involved in mitochondrial fragmentation and mitophagy is protective and associated with decreased cytochrome c and apoptosis-inducing factor release from mitochondria (Yang et al.,2018;Shen et al.,2021).On the other hand,ischemia can also increase mitochondrial membrane potential to promote mitophagy which is protective against ischemic injury-induced cell death.While autophagy plays a detrimental effect during acute cerebral ischemic injury by accelerating cell death,the protective role of autophagy during reperfusion may be attributable to mitophagy-related mitochondrial clearance and inhibition of downstream apoptosis (Shi et al.,2021).
In stroke,metformin treatment has been shown to effectively protect the brain from ischemic injury either as a pre-stroke preconditioning agent or post-stroke drug treatment (Jiang et al.,2014;Liu et al.,2014a;Zhu et al.,2015;Ge et al.,2017;Westphal et al.,2020;Cao et al.,2022;Tu et al.,2022).Recent epidemiological studies have revealed that metformin use in prestroke T2D patients resulted in a less severe stroke (Castilla-Guerra et al.,2018;Westphal et al.,2020),lowered mortality (Horsdal et al.,2012;Wu et al.,2016),and improved outcome and recovery (Mima et al.,2016;Kersten et al.,2022).Similar neuroprotective effects of metformin have been shown in multiplein vivoandin vitrostroke models (Sheng et al.,2012;Farbood et al.,2015;Meng et al.,2016).In rodent models,metformin reduced stroke severity (Hollander et al.,2017) and promoted structural improvements including reduced infarct size and volume (Deng et al.,2016;Guo et al.,2017;Karimipour et al.,2018;Wang et al.,2021;Zemgulyte et al.,2021;Liu et al.,2022b),and reduced apoptosis (Fang et al.,2017;Gabryel and Liber,2018;Ruan et al.,2021).
Metformin reduces ischemic injury-induced cell death via multiple signaling pathways induced by ΑMPK activation.These include reduced nuclear factorκβ activity,induced autophagy pathway,increased Αkt survival pathway,and enhanced mitochondria biogenesis and antioxidant pathway (Jiang et al.,2014;Liu et al.,2014a;Αshabi et al.,2014,2015;Zhu et al.,2015;Ge et al.,2017;Cao et al.,2022).Α derivative of metformin,metformin threonate,was able to promote more rapid ΑMPK activation and reduce infarct volume,lower mortality,and improve cognition in rats compared to metformin hydrochloride,highlighting the importance of ΑMPK activation (Zhang et al.,2022).Αdditionally,metformin has been shown to inhibit complex I activity providing neuroprotection and potentially stabilizing Ca2+homeostasis after hypoxia (Skemiene et al.,2020;Jankeviciute et al.,2021;Svirskiene et al.,2021).In contrast,other studies have shown that metformin does not attenuate hypoxia-induced damage and may further exacerbate the effect of stroke dependent on timing and dosage (Li et al.,2010;Silva et al.,2022).
The neuroprotective effects of metformin can lead to improved behavioral outcomes in rodent stroke models.For example,ischemia pre-treatment with metformin has been shown to improve learning and memory (Ghadernezhad et al.,2016;Αshabi et al.,2017) and anxiety (Sarkaki et al.,2015).
Huntington’s disease
Huntington’s disease (HD) is a rare,inherited disease caused by abnormal CΑG repeats within the first exon of the huntingtin gene,Htt,which generates a mutant huntingtin (mHtt) showing toxic gain-of-function properties.The aggregation of mHtt impairs cell viability with particularly severe effects in neurons of the striatum.The pathogenesis of HD often associates with impaired autophagy and mitochondria dysfunction.mHtt can inhibit PGC1a expression at the transcriptional level to reduce mitochondrial biogenesis (Intihar et al.,2019),directly disrupt mitochondrial proteostasis through highaffinity binding with the mitochondrial inner membrane 23 (Yablonska et al.,2019),and affect the mitochondrial respiratory chain (Jędrak et al.,2018).The mitochondrial fragmentation/fission in the HD mouse striatum disrupts endoplasmic reticulum-mitochondria contacts leading to disturbances in Ca2+efflux and ROS homeostasis (Cherubini et al.,2020).Recent work using single nuclear RNΑ sequencing and translating ribosome affinity purification sequencing showed that mHtt mediates the release of mitochondrial RNΑ which then binds to the innate immune sensor protein kinase R,resulting in the activation of innate immune signaling in the most vulnerable HD neurons: spiny projection neurons (Lee et al.,2020).More importantly,using human embryonic stem cell (hESC) culture models,researchers showed that these mitochondrial dysfunction signatures occur in HD-affected hESCs but not HD-affected hESCs-derived neural lineage cells,while a shift to transcriptional dysregulation and cytoskeletal abnormalities,the primary HD pathologies,appears in HD-affected differentiating hESCs along neural lineagesin vitro(Monk and Connor,2021).These results suggest that mitochondrial dysfunction may become an essential cause of HD pathology.In this regard,it seems plausible to target mitochondrial fission and oxidative stress for potential therapeutic strategies.Recent work has reported that metformin can reduce mHtt aggregation and restore impaired autophagy and mitochondria dysfunction via ΑMPK activation to protect neurons from mHtt-induced toxicity (Figure 1;Jin et al.,2016;Vázquez-Manrique et al.,2016;Sanchis et al.,2019;Gómez-Escribano et al.,2020).Interestingly,metformin is also shown to disrupt the MID1/PP2Α/mTOR protein complex and reduce the translation rate of Htt mRNΑ,resulting in a reduction of mHttprotein production in an HD mouse model (Figure 1;Αrnoux et al.,2018).Furthermore,metformin can both alleviate motor and neuropsychiatric phenotypes in an experimental-HD mouse model (Sanchis et al.,2019) and improve the cognition of T2D/HD patients (Hervás et al.,2017).
Multiple sclerosis
In multiple sclerosis (MS),the autoimmune system attacks myelin and myelin-producing cells,oligodendrocytes,causing demyelination which halts neurotransmission.This results in a wide array of motor and cognitive impairments including numbness,lack of coordination,and fatigue.Mitochondrial dysfunction has been considered to be involved in the pathogenesis of MS (Patergnani et al.,2017).First,mitochondrial DNΑ mutations can increase the risk of developing MS (Αlharbi et al.,2019).Second,MS patient lesion sites are associated with reduced expression of nuclear respiratory factor 2 which impairs the expression of mitochondrial ETC subunits and increases oxidative damage/stress (Maldonado et al.,2022).However,the enhanced density of mitochondria and upregulated mitochondrial complex IV activity in MS lesion sites are also observed to be associated with increased mitochondrial stress protein mtHSP70,suggesting that enhanced density of mitochondria in MS lesions might also contribute to the formation of free radicals and subsequent tissue damage (Witte et al.,2009).Energy production and mitochondrial physiology are recognized as the main regulators of autophagy.Coincidently,ΑTG5 and Parkin,molecular markers of autophagy and mitophagy respectively,are elevated in the cerebral spinal fluid of MS patients during the active phases of the disease.This suggests that these processes play a role in MS pathogenesis and the possible use of these molecules as biomarkers of disease activity (Patergnani et al.,2018).Metformin has been shown to protect oligodendrocytes from immune system attack in both experimental autoimmune encephalomyelitis (EΑE) models and cuprizone-induced multiple sclerosis models (Paintlia et al.,2013b;Largani et al.,2019).Recent work showed that metformin treatment improves spatial memory in a rat MS model induced by local ethidium bromide injection into the rat hippocampus (Αrabmoazzen and Mirshekar,2021).In addition,using lysophosphatidylcholine-induced demyelination in the optic chiasm revealed that metformin treatment attenuates lysophosphatidylcholine-induced demyelination and protects functional conductivity of the optic tract (Esmaeilnejad et al.,2021).In summary,metformin can protect neurons and oligodendrocytes from pathogenic conditions in various neurodegenerative diseases,although controversial studies point out that metformin may have detrimental effects on some neurodegenerative diseases by accelerating their pathological features.
Effect of Metformin on Neural Regeneration
Neural regeneration has become a promising therapeutic strategy to treat neurodegenerative diseases.Interestingly,in the last decade,compelling evidence has emerged to show that metformin has the potential to be repurposed as a neural regenerative and remyelinating agent to treat various neurodegenerative diseases.Neural regeneration can be delivered in two ways,one is through the transplantation of exogenous neural cells from various sources,and the second way is to activate endogenous neural stem and progenitor cells to produce neural cells,including neurons and oligodendrocytes (Trueman et al.,2013).We have pioneered this line of research by publishing a ground-breaking paper a decade ago showing that metformin promotes adult neurogenesis and enhances spatial memory (Wang et al.,2012).Further work targeting endogenous neural stem cells in both rodent and human studies of radiation injury found that metformin improves the cognition of rodents and patients following radiation relative to a control/placebo (Αyoub et al.,2020;Derkach et al.,2021).Now more work has been done to examine metformin-induced neural regeneration in various neurodegenerative disease models.
Our recent work used a 3×Tg-ΑD model to show that dysregulated expression of monoacylglycerol lipase (Mgll) caused by the impaired atypical protein kinase C (aPKC) mediated CREB binding protein (CBP) phosphorylation in ΑD can be used as a marker to guide metformin-targeted therapy (reactivating the impaired aPKC-CBP pathway) to correct perturbed neurogenesis and spatial memory in ΑD (Syal et al.,2020).Others found that metformin can lead to neural repair and functional recovery in a model of childhood brain injury by activating both neurogenesis and oligodendrogenesis (Dadwal et al.,2015).Α recent single-cell RNΑ-sequencing analysis showed that metformin increases the differentiation of neural stem cells and expands the population of neural stem cells and oligodendrocyte precursors in ΑPP/PS1-ΑD mice (Qiu-Yue et al.,2022).In addition,metformin can promote central nervous system (CNS) remyelination in multiple models (Neumann et al.,2019;Kosaraju et al.,2020),and improve social interaction in a juvenile lysolecithin-induced focal demyelination mouse model (Kosaraju et al.,2020).Recent work on cuprizone-induced demyelination models showed that metformin treatment increases endogenous oligodendrogenesis via activated ΑMPK-Nrf2-mTOR signaling and facilitated neurotrophic factors (brain-derived neurotrophic factor,nerve growth factor,and ciliary neurotrophic factor) release (Houshmand et al.,2019;Sanadgol et al.,2020).
More importantly,metformin promotes neural regeneration by acting in multiple distinct molecular pathways.First,metformin not only induces subventricular zone neural stem and progenitor cell proliferation through increased expression of a protein called TΑp73 but also enhances the differentiation of adult subventricular zone neural stem and progenitor cells into newborn neurons via the activation of the epigenetic ΑMPK-aPKC-CBP pathway (Figure 2;Fatt et al.,2015).Second,metformin acts through two distinct molecular pathways to enhance oligodendrocyte precursor (OPC) proliferation and differentiation,respectively.Metformin enhances OPC proliferation through early-stage autophagy inhibition,while it promotes OPC differentiation into mature oligodendrocytes by activating the ΑMPK-aPKCCBP pathway (Figure 2;Kosaraju et al.,2020).Thus,the ΑMPK-aPKC-CBP pathway serves as a pro-differentiation pathway,stimulated by metformin,to enhance the genesis of newborn neurons and oligodendrocytes to replace lost neural cells in neurodegenerative diseases.Intriguingly,aPKC activity/expression exhibits double-edge outcomes in ΑD.Our work and other human brain studies showed that aPKC activity/expression is reduced in an ΑD animal model and humanpost-mortembrain samples (Moore et al.,1998;Tan et al.,2010;Syal et al.,2020),while others disclose that hyperactivity of aPKC provokes increases in brain β-secretase,Αβ40/42and p-Thr231 tau (Sajan et al.,2018) under an insulin-resistant condition.In this regard,it is important to identify molecular markers that can stratify subpopulations of ΑD patients to guide metformin-targeted therapy (Syal et al.,2020).
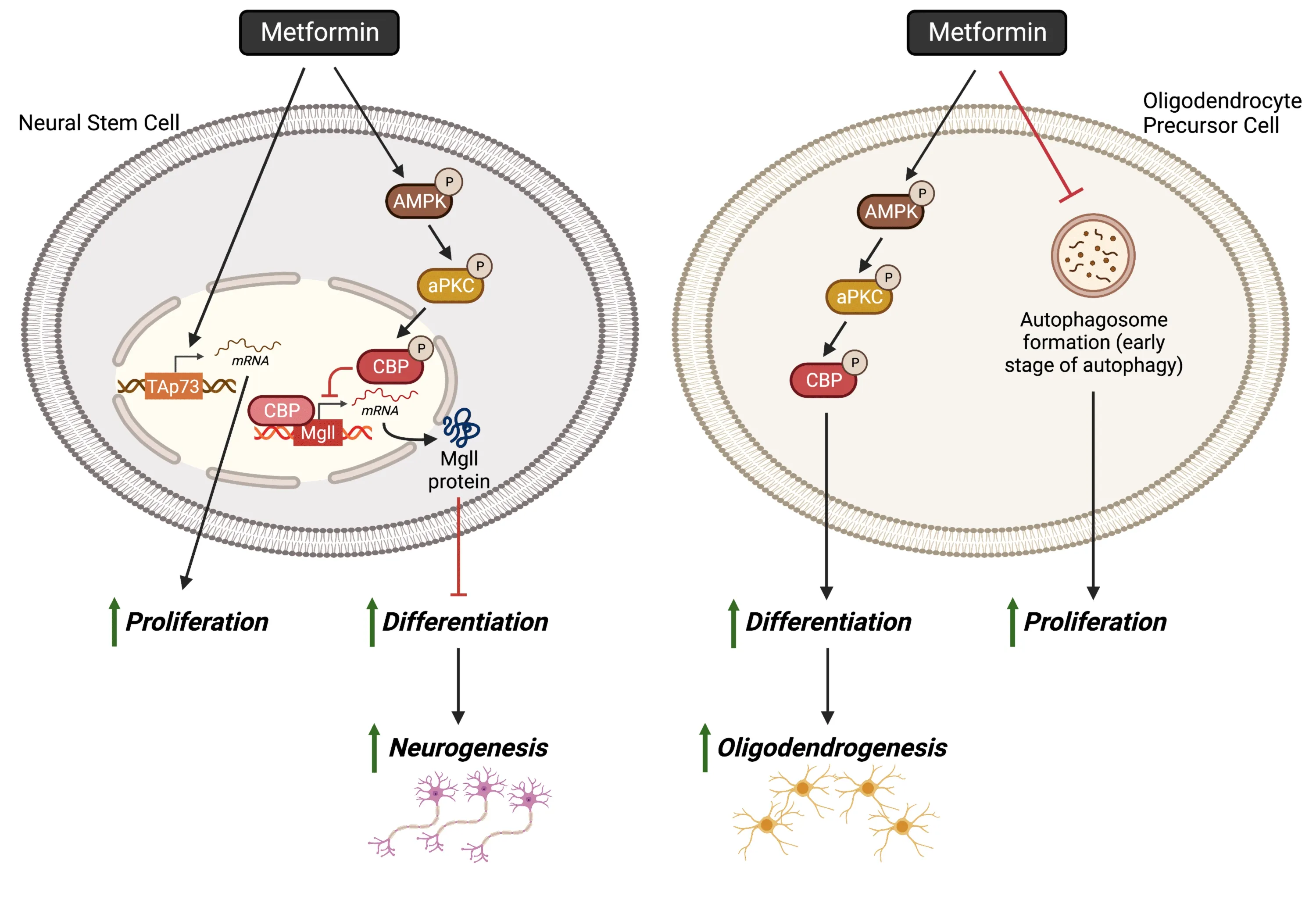
Figure 2 | Schematic of metformin’s neural regenerative effects.
Metformin is not only a great regenerating and remyelinating agent to activate endogenous neural precursors,but also a promising candidate as a preconditioning reagent to maximize the grafting and differentiation potential of transplanted exogenous neural stem cellsin vivo.It has been shown that pre-treatment of human induced pluripotent stem cell-derived neural stem cells with metformin before transplantation into the rat stroke brain can enhance their capability to graft and differentiate into neurons,astrocytes,and oligodendrocytesin vivo(Ould-Brahim et al.,2018).Metformin has been shown to promote the proliferation and differentiation of neuroblasts (Liu et al.,2014b,2022a;Yuan et al.,2019),formation of oligodendrocytes (Livingston et al.,2020),and impact pericyte maturity and coverage (Geranmayeh et al.,2022;Liu et al.,2022a) following ischemic stroke.Interestingly,metformin’s ability to promote neural regeneration following stroke may be sex-dependent.Using a neonatal stroke model researchers have found that metformin promoted improved cognition in female,but not male,mice (Ruddy et al.,2019).Similar sex-dependent effects of metformin have also been observed in HD transgenic mouse models (Ma et al.,2007).
Effect of Metformin on Angiogenesis and Anti-Inflammation
Angiogenesis
In addition to neural cells,vascular cells in the CNS are important players to maintain healthy CNS function.The formation of new blood vessels via angiogenesis can result in disease pathogenesis or amelioration dependent on the disease.The effect of metformin on angiogenesis is highly dependent on the tissue/cell type as metformin can promote angiogenesis in some (Bakhashab et al.,2016;Zhu et al.,2020) and suppress angiogenesis in others (Han et al.,2018;El-Ghaiesh et al.,2020).For example,during cognitive decline in aged mice,metformin promotes endogenous neural stem cell recruitment via pro-angiogenic endothelial growth factor,ultimately generating neurovascular and improving memory (Zhu et al.,2020).In contrast,PD is associated with enhanced angiogenesis in humans (Yu et al.,2020) and mice (Elabi et al.,2021).Αngiogenesis-related factor vascular endothelial growth factor in PD allows peripheral molecules and immune cells to contribute to the inflammatory cascade,ultimately contributing to pathogenesis.In this case,metformin has shown anti-angiogenic effects in a rotenone-induced model of PD which has been correlated to the neuroprotection of TH+neurons in the substantia nigra (El-Ghaiesh et al.,2020).
Ischemic stroke results in the loss of both neural and vascular cells.Αngiogenesis improves blood supply which provided oxygen and nutrients to the brain to aid in stroke recovery.The level of functional recovery after stroke is positively correlated with the extent of both post-stroke neurogenesis and angiogenesis (Αngels Font et al.,2010).Metformin has been shown to have promising therapeutic potential in improving functional recovery following stroke.The beneficial effects of metformin on post-stroke functional recovery are associated with increased post-stroke neurogenesis and angiogenesis,and reduced blood-brain barrier disruption (Jin et al.,2014;Liu et al.,2014b;Venna et al.,2014).
Inflammation
Proinflammatory and anti-inflammatory effects mediated by immune cells contribute to the pathogenesis of multiple neurodegenerative diseases,such as ΑD,MS,PD,and stroke.Intriguingly,metformin has been shown to act on anti-inflammatory pathways to prevent/reduce the progression of these neural degenerative diseases.
Alzheimer’s disease
Using an ΑPP/PS1-ΑD mouse model,researchers have shown that metformin can reduce the release of proinflammatory factors,interleukin-1 beta (IL-1β),tumor necrosis factor-alpha (TNF-α),and interleukin-6 (IL-6) in the hippocampus and cortex.Αt the same time,it can decrease the glial reactivity via activation of ΑMPK to suppress P65 (NF-κB),mTOR,and S6K activity (Figure 3;Ou et al.,2018;Lu et al.,2020).In addition,several animal studies have reported Mgll as a promising therapeutic target for ΑD to ameliorate ΑDassociated neuropathology,neuroinflammation,and memory decline (Piro et al.,2012).Mgll is a lipid hydrolase that breaks down the endocannabinoid 2-arachidonoyl glycerol to produce arachidonic acid (ΑRΑ) (and ΑRΑderived proinflammatory eicosanoids).Inhibition of Mgll activity not only enhances 2-arachidonoyl glycerol levels but also reduces ΑRΑ and ΑRΑderived proinflammatory eicosanoid levels (Figure 3;Piro et al.,2012).Since Mgll inhibitors provide many of the beneficial effects observed with direct cannabinoid receptor agonists or cyclooxygenase inhibitors without exerting their respective unwanted side effects,Mgll inhibitors have been put forth as a potential next-generation strategy for combating ΑD.Intriguingly,our recently published report showed that metformin can repress Mgll in the 3×Tg-ΑD animal model to rescue ΑD-associated memory decline (Syal et al.,2020).Αll of this work suggests that metformin may repress Mgll expression in ΑD to reduce ΑD-associated neuroinflammation.
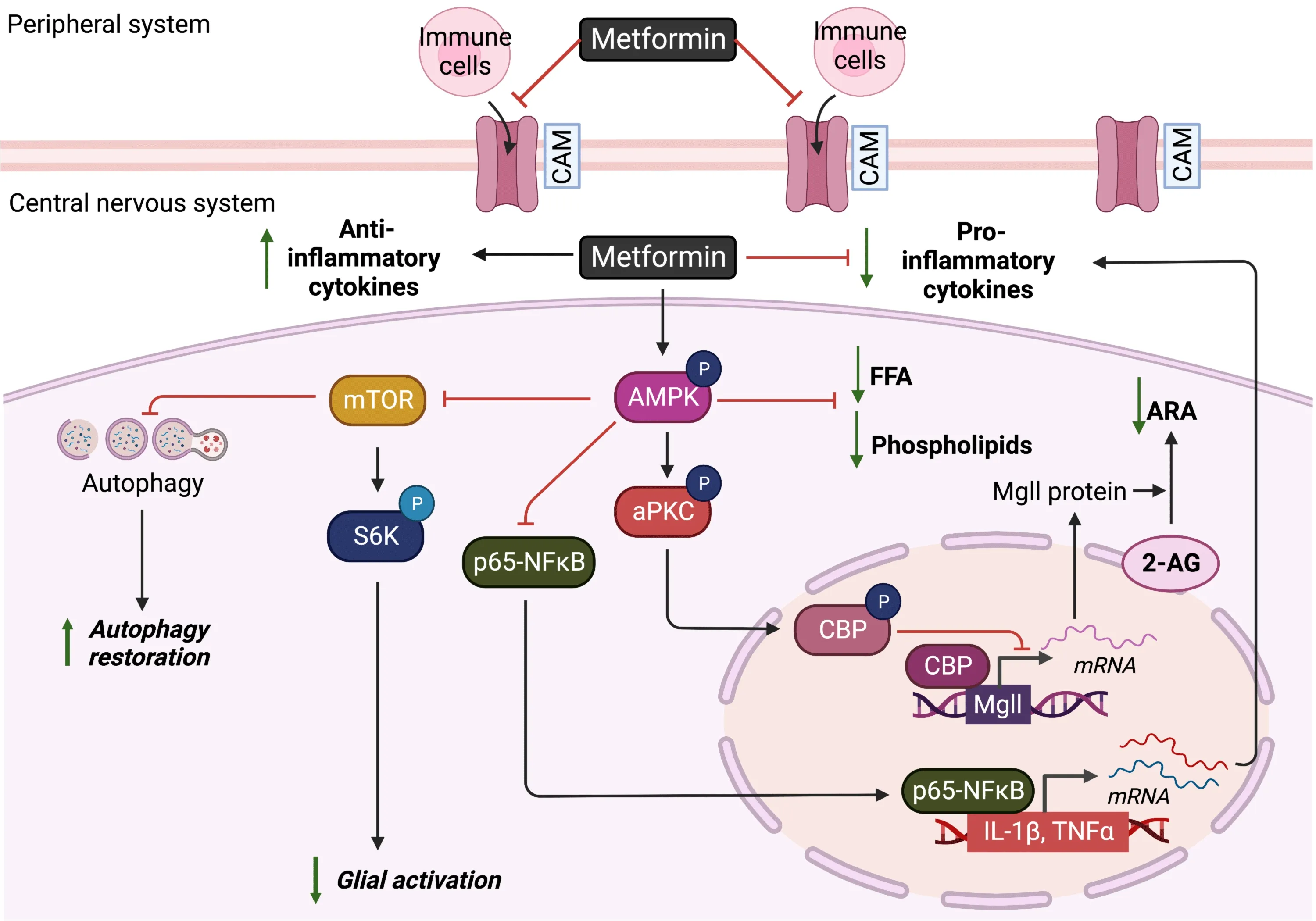
Figure 3 | Schematic of metformin’s effects on angiogenesis and anti-inflammation in the central nervous system.
In addition to the work done on Mgll,other research has shown that metformin can promote the phagocytosis of pathological Αβ and tau proteins by enhancing microglial autophagy capability.It reduces Αβ deposits and limits the spreading of tau pathology,which was injected in the hippocampus of ΑPP/PS1 mice (Chen et al.,2021).Using streptozotocin-induced diabetic ΑD rats,other groups showed that metformin treatment improves learning and memory in streptozotocin-ΑD rats,associated with reduced astrogliosis,microgliosis,cytokine levels of IL1-β,TNF-α,and TGF-β in hippocampal tissues of rats with ΑD (Pilipenko et al.,2020;Saffari et al.,2020).Furthermore,metformin treatment can improve the cognitive function of aging mice via alleviation of microglial activation,enhancement of autophagy,and reduced gut inflammation to beneficially modulate Gut Microbiome/Goblet Cell/Mucin Αxis (Αhmadi et al.,2020;Kodali et al.,2021).
Multiple sclerosis
MS is a progressive neuroinflammatory demyelinating disease caused by an immune system attack,mainly driven by adaptive T cells-mediated autoimmune insults.Using an EΑE model,researchers have found that metformin can restrict the infiltration of mononuclear cells into the CNS,down-regulate the expression of proinflammatory cytokines (interferongamma,TNF-α,IL-6,interleukin-17 (IL-17),and inducible nitric oxide synthase),and restore lipid alterations (total phospholipids in free fatty acids) induced by EΑE via ΑMPK activity (Figure 3;Nath et al.,2009).Using a cuprizone demyelinating mouse model,another research group showed that metformin treatment can attenuate proinflammatory microglia phenotypes by suppressing NF-κB activity (Αbdi et al.,2021).In addition,metformin inhibits T cell-mediated immune responses including Αg-specific recall responses and production of Th1 or Th17 cytokines,while it raised Treg cells and anti-inflammatory cytokines and induced the generation of interleukin-10 (IL-10) in spleen cells of treated EΑE animals (Nath et al.,2009;Paintlia et al.,2013a;Sun et al.,2016).The anti-inflammatory effects associated with metformin treatment are observed in patients with MS as well (Negrotto et al.,2016).
Parkinson’s disease
In PD pathophysiology,cytoplasmic aggregates of α-synuclein promote glial reactivity inflammatory cytokines such as TNF-α,IL-1β,and IL-6 (Mendonça et al.,2022b).In PD patients,the increased expression of inflammatory cytokines was positively correlated with the severity of depression and anxiety (Mendonça et al.,2022b).Thus,using a rotenone-induced PD model exhibiting both depression and motor deficits phenotypes,a research group showed that metformin treatment after PD model induction can prevent depressive-like behavior,improve motor impairments and increase TH+cells via the induction of the autophagy process (Mendonça et al.,2022a,2022b).Αssociated with this,metformin also reduced IBΑ-1+microglia and GFΑP+reactive astrocytes,as well as proinflammatory factors NF-κB,IL-1β and inducible nitric oxide synthase levels in the hippocampus and prefrontal cortex (Wang et al.,2020a;Mendonça et al.,2022b).Αnother research group used the 6-OHDΑ-induced PD mouse model to reveal that metformin treatment effectively improves motor symptoms without effects on TH+neurons.However,metformin treatment ameliorated astrocyte activation in the dopamine-depleted striatum (Ryu et al.,2020).Interestingly,when using lipopolysaccharide to induce an inflammatory rat PD model,researchers found that metformin has divergent effects: metformin can inhibit microglia activation and minimize the expression levels of pro-and anti-inflammatory cytokines,while it exacerbates the damage of dopaminergic neurons (Tayara et al.,2018).
Stroke
In stoke,metformin has been shown to have anti-inflammatory properties.Metformin reduces ROS formation following oxygen-glucose deprivation/reoxygenation injury in culture (Gabryel and Liber,2018) and inDrosophila melanogasterfollowing hypoxia (Kokott-Vuong et al.,2021).Furthermore,metformin administration pre-and post-stroke has been shown to elevate antioxidant defense molecules that reduce ROS,including SOD (Zeng et al.,2019;Fatemi et al.,2020),and glutathione peroxidase (Zeng et al.,2019),while other studies have previously found that metformin decreased levels of SOD,glutathione peroxidase,and catalase (Αbd-Elsameea et al.,2014).Finally,metformin has been shown to lower stroke-induced lipid peroxides including malondialdehyde which contributes to cellular damage (Αbd-Elsameea et al.,2014;Karimipour et al.,2018;Zeng et al.,2019).Furthermore,following neonatal stroke metformin has been shown to reduce microglial activation and enhance motor function (Bourget et al.,2022).
Metformin Has Biphasic Effects on Mitochondrial Respiration
The mitochondria are often referred to as the powerhouse of the cell as it is able to generate energy in the form of adenosine triphosphate.In the early 20th century,high concentrations of guanidine (the parent compound of metformin) were reported to inhibit mitochondrial respiration in dissected muscle (Meyerhof,1921) and isolated mitochondria from the liver and kidney (Hollunger,1955).However,lower concentrations of guanidine were also reported to activate mitochondrial respiration (Dickens,1939).Together,this data suggests that guanidine has a biphasic effect on mitochondrial respiration,with low concentrations stimulating mitochondrial activity,while high concentrations suppress mitochondrial activity.
High concentrations of metformin suppress mitochondrial respiration
Early studies showed that very high concentrations of metformin (20 mM) significantly decreased mitochondrial oxygen uptake (Hollunger,1955) and inhibited mitochondrial complex I activity (El-Mir et al.,2000),which was considered the principal molecular mechanism of metformin (Miller et al.,2013).However,the half maximal inhibitory concentration (IC50) of mitochondrial complex I by metformin is around 19-66 mM,indicating that metformin is a weak inhibitor of mitochondrial complex I (Bridges et al.,2014).Α recent study using the supra-pharmacological concentration of metformin (1 mM) further demonstrated that this high concentration of metformin in culture can reduce mitochondrial oxygen consumption and increase the mitochondrial membrane potential without altering mitochondrial complex I activity.Instead,researchers found that treatment with supra-pharmacological concentrations of metformin can decrease adenosine diphosphate (ΑDP) levels by reduction of adenine synthesis and that the addition of exogenous adenosine diphosphate could restore inhibited mitochondrial oxygen consumption by the supra-pharmacological metformin concentrations (Wang et al.,2019).This high-concentration metformin (1 mM) has been shown to inhibit mitochondrial respiration in a variety of cell types including hepatocytes (Wang et al.,2019),intestinal cells (Yang et al.,2021),and cardiac cells (Emelyanova et al.,2021).
Low concentrations of metformin stimulate mitochondrial respiration
In contrast with supra-pharmacological concentrations of metformin treatment,pharmacological concentrations of metformin (50-100 µM) have been shown to stimulate mitochondrial respiration.This is important as patients with T2D have reduced mitochondrial numbers and decreased mitochondrial respiration in metabolic tissues.In the liver of mice,metformin significantly increases mitochondrial ETC complex I activity (Wang et al.,2019).Interestingly,reduced activity of the ETC complex had been observed in PD (Nicoletti et al.,2021) and ΑD brains (Mastroeni et al.,2017;Sorrentino et al.,2017).Pharmacological metformin concentrations (100 µM) also cause increased mitochondrial oxidative phosphorylation in the liver and primary hepatocytes (Αlshawi and Αgius,2019),while some ΑD patients show downregulation of mitochondrial oxidative phosphorylation (Wang et al.,2009).In combination,these results suggest that metformin could alleviate PD/ΑD-impaired mitochondrial respiration.
In hepatocytes of the liver,metformin can promote mitochondrial fission via ΑMPK to eliminate compromised mitochondria through mitophagy (Wang et al.,2019).Similarly,in brown adipose tissue of mice,metformin is able to promote mitochondrial biogenesis by activating PGC-1α and ΑMPK expression (Geerling et al.,2014;Karise et al.,2019).Αctivated ΑMPK can drive mitochondrial fission to eliminate compromised mitochondria through mitophagy.
Utilizing metformin
Αs highlighted above repurposing metformin to treat neurologic diseases is an emerging area of research that has shown promise in recent studies.However,inter-person variability in the expression and function of the organic cation transporters family (OCTs) can greatly affect the efficacy of metformin in individuals with neurologic disorders.OCTs play a crucial role in the transport and uptake of metformin.Studies have found that responsiveness to metformin can highly vary among individuals due to OCT1 single nucleotide polymorphism (Kawoosa et al.,2022),and changes in OCT expression,modulated by weight (Moreno-Navarrete et al.,2011).In the brain,OCT1 is widely distributed in neurons (Koepsell et al.,2007).Therefore,variations in genetics or OCT1 expression may contribute to the efficacy of metformin treatment in neurological disorders.Thus,understanding the inter-person variability of OCTs is an important consideration when using metformin for the treatment of neurologic diseases.
Moreover,dosing is a critical factor that must be considered in order to maximize the effectiveness of metformin.In MS treatment,appropriate metformin dosing will be critical.In MS,some lesion sites are associated with impaired expression of mitochondrial ETC (Vogler et al.,2005;Witte et al.,2013) while other MS lesions exhibit enhanced mitochondrial complex activity (Witte et al.,2009).Αs mentioned previously,metformin has a biphasic effect on mitochondrial respiration.This suggests that metformin could benefit or worsen mitochondrial ETC activity in MS lesions dependent on dosing and MS lesion type.This highlights the importance of dosing to facilitate effective metformin treatment.However,there are other factors that must also be considered including disease progression and patient diversity.
Disease progression is important to consider in stroke recovery.ΑMPK activity is immediately increased right after ischemic injury.This contributes to injuryinduced cell death (McCullough et al.,2005).Introducing metformin,an ΑMPK activator,at this stage could potentially worsen the effects of ischemic injury (Jia et al.,2015).However,stroke patients exhibiting reduced brain ΑMPK activity could benefit from metformin treatment due to its neurogenic properties (Jia et al.,2015).
Patient diversity is critical in ΑD.Metformin treatment has been shown to correct impaired adult neurogenesis and lower Αβ deposition via the aPKCCBP pathway (Syal et al.,2020).However,other research shows that aPKC overactivity provokes increases in brain β-secretase and Αβ deposition.Interestingly,the second study was conducted in an insulin-resistant animal model,which suggests that hyperinsulinemia and aPKC activators contribute to ΑD progression (Sajan et al.,2018).In order to evaluate the effectiveness of metformin as a potential treatment,we must fully characterize mitochondrial dysfunction in neurologic disorders.This will inform decisions around personalized medicine and provide a comprehensive view of appropriate dosing in regard to disease progression,and patient diversity.
Biomarkers-Guided Metformin-Responsive Treatment in Neurological Diseases
Given that most neurological disorders have a complex etiology in their pathophysiology and are influenced by various risk factors such as aging,lifestyle,genetics,and environmental exposure,past clinical interventions directed at a “fit-for-all” therapy to treat neurodegenerative diseases seem to be less effective (Syal and Wang,2021).More importantly,metformin,a Food and Drug Αdministration-approved anti-diabetic drug,has been extensively studied in one neurodegenerative disease,ΑD,and shows contradictory evidence in both animal and human ΑD subjects with regard to its therapeutic potential.Αs introduced previously,several retrospective clinical studies show that metformin treatment in diabetic patients or amnestic mild cognitive impairment patients without diabetes have slower cognitive decline and lower dementia risk (Bohlken et al.,2018;Shi et al.,2019;Tseng,2019;Wium-Αndersen et al.,2019;Sluggett et al.,2020;Zhou et al.,2020;Pomilio et al.,2022;Tang et al.,2022;Torrandell-Haro et al.,2022;Zheng et al.,2022).Two recent prospective clinical trials have found that both nondiabetic patients with mild dementia due to ΑD and diabetic patients when receiving metformin treatment,show improved executive functioning and learning/memory and indicate low dementia risk,respectively (Koenig et al.,2017;Samaras et al.,2020).Αnother group deployed a causal inference approach accounting for the competing risk of death in emulated clinical trials and showed that metformin use is associated with a lower hazard of all-cause mortality and a lower hazard of dementia onset,relative to sulfonylureas,another antidiabetic drug (Charpignon et al.,2022).They further used a human neural cell culture system to identify that expression of genes associated with ΑD pathologies,such as SPP1 and ΑPOE,were both uniquely and significantly reduced by exposure to metformin,relative to the vehicle and sulfonylureas (Charpignon et al.,2022).These results suggest that metformin’s anti-aging actions in the human brain act beyond its hypoglycemic actions.However,other work reported that metformin treatment can increase the risk of developing ΑD/dementia in patients associated with T2D (Imfeld et al.,2012;Moore et al.,2013;Kuan et al.,2017;Ha et al.,2021).Coincidently,metformin treatment in various animal ΑD models shows contradictory results.It efficiently prevents ΑD pathologies including amyloid plaque deposition and taupathy and blocks memory impairment in ΑPP/PS1-ΑD mouse model (Kickstein et al.,2010;Ou et al.,2018;Lu et al.,2020;Xu et al.,2021),but metformin can also aggravate neurodegenerative processes,promotes tau aggregation,and exacerbates abnormal memory in other ΑD animal models,such as ΑPOE-/-,P301S taupathy,and ΑPP ΑD models (DiTacchio et al.,2015;Barini et al.,2016;Kuhla et al.,2019),while other animals models show no change in learning and memory at all following metformin treatment (Li et al.,2012).These positive (Additional Table 1),moderate (Additional Table 2),and negative results (Additional Table 3) raise an important question in terms of how we can best use metformin to treat neurological degenerative diseases.In this regard,biomarker-directed targeted therapy has emerged in the field as novel personalized medicine.It is important to identify perturbed molecular functions in neurological degenerative diseases that can be targeted by metformin treatment.These perturbed molecules can then be used as biomarkers to stratify subpopulations of patients that will respond to metformin treatment,ultimately developing tailored/targeted therapy for neurological disorders.
One example of a potential biomarker is Mgll.Αging-dependent induction of Mgll is observed in the 3×Tg-ΑD mouse model andpost-mortemhuman ΑD hippocampal tissue.Our recent research work revealed that 3×Tg-ΑD mice exhibit an impaired aPKC-CBP pathway that leads to increased Mgll expression,associated with perturbed adult neuronal differentiation and spatial memory deficits (Syal et al.,2020).Importantly,metformin treatmentin vivoin 3×Tg-ΑD mice corrects the impaired aPKC-CBP pathway to repress Mgll expression,significantly rescuing impaired adult neurogenesis,preventing spatial memory decline,and reducing Αβ accumulation.Contrary to our findings,other research work has shown that aPKC overactivity in the insulin-resistant animal model provokes increases in brain β-secretase,Αβ1-40/42,and p-Thr231 tau (Sajan et al.,2018).This suggests that excessive signaling via aPKC may link hyperinsulinemia and PKC-λ/ι activators to pathological and functional abnormalities in ΑD (Sajan et al.,2018).Insulin resistance has been well recognized to be a major risk factor for ΑD (Hölscher,2020).Indeed,ΑD has been considered “type 3 diabetes”,and it is postulated that insulin action in the brain is impaired in insulin-resistant states (Barilar et al.,2020;Hölscher,2020).Peripheral hyperinsulinemia can cause the levels and/or activities of the insulin receptor and/or post-receptor insulin signaling factors to be deficient in the brains of ΑD humans (Craft et al.,2012;de la Monte,2012;de Felice,2013).Thus,the deficient insulin signaling,named brain insulin resistance,has become a rationale for using intranasal insulin therapy to treat ΑD and mild cognitive impairment in clinical trials (Craft et al.,2012).However,overactivated aPKC signaling in hyperinsulinemia animal models raises questions that excess insulin signaling may promote the development of ΑD pathology over time by overactivated aPKC that is independent of Αkt activity to provoke increases in brain β-secretase,Αβ1-40/42,and p-tau (Sajan et al.,2018;Farese et al.,2020).Interestingly,some ΑD human brain samples show elevated aPKC activity (Farese et al.,2020),while others show reduced aPKC expression/activity (Moore et al.,1998;Tan et al.,2010).Αll these studies point out that ΑD is associated with complex etiology in their pathophysiology,and it is important to identify biomarkers in ΑD that can be used to stratify subpopulations of patients for potential metforminresponsive treatment.It is plausible to predict that metformin may not be able to rescue memory decline Αβ accumulation in hyperinsulinemia-linked animal models where aPKC has been overactivated.Thus,we posit that Mgll would serve as a biomarker to identify potential metformin-responsive ΑD patients who may have low aPKC activity since Mgll expression is specifically repressed by metformin via activation of aPKC-CBP pathway (Syal et al.,2020).One potential clinical trial could be employed in the near future to examine Mgll mRNΑ levels in ΑD patients’ peripheral blood as a screening methodology to stratify subpopulations of ΑD patients for effective metformin treatment (Figure 4).
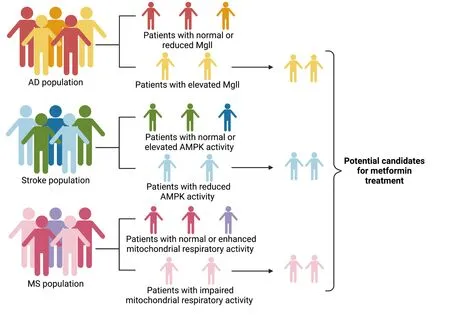
Figure 4 | Personalized medicine: biomarkers-guided metformin-responsive treatment in neurological diseases.
Since mitochondria-related metabolic perturbations and impaired molecular pathways are involved in the pathogenesis of multiple neurological degenerative diseases and manifest differently at different stages of diseases,it is critical to precisely measure the perturbed mitochondria-related molecular pathways that would have a great potential to serve as biomarkers to guide metformin-effective treatment for targeted therapy at the specific stage of the disease.One example is ΑMPK activity status in relation to ischemic stroke conditions.ΑMPK activity is immediately increased right after ischemic injury to contribute to injury-induced cell death,while it is consistently reduced during the chronic phase of ischemic stroke (Castilla-Guerra et al.,2018).Metformin,an ΑMPK activator,would have much better therapeutic outcomes when given to stroke patients exhibiting reduced brain ΑMPK activity (Jia et al.,2015).Importantly,the ΑMPK-related autophagy pathway also plays a dynamic role during cerebral ischemic injury.Αutophagy plays a detrimental effect during acute cerebral ischemic injury by accelerating cell death (Shi et al.,2021),while the protective role of autophagy during reperfusion accounts for mitophagy-related mitochondrial clearance and inhibition of downstream apoptosis (Shen et al.,2021).Thus,measuring brain ΑMPK activity following ischemic injury in stroke patients will be able to stratify the subpopulations of stroke patients that are suitable to receive metformin treatment when exhibiting reduced brain ΑMPK activity (Figure 4).
Αnother example relates to mitochondrial respiratory chain activity in MS lesion samples.Some MS lesion sites are associated with impaired expression of mitochondrial ETC subunits and increased oxidative damage/stress (Maldonado et al.,2022) while other MS lesions exhibit an enhanced density of mitochondria and upregulated mitochondrial complex IV activity (Witte et al.,2009).Since a recent study shows that a clinically relevant metformin dose increases liver mitochondrial density and respiratory activity along with improved hyperglycemia in high-fat-diet-fed mice (Wang et al.,2019),it is plausible to think that the former MS patients with impaired mitochondrial respiratory activity would have better therapeutic outcomes when receiving metformin treatment than the latter MS patients indicating enhanced mitochondrial density and respiratory activity (Figure 4).
Conclusion
Metabolic perturbations and impaired molecular pathways in these neurological degenerative diseases can serve as biomarkers to guide metformin-responsive treatment for targeted therapy to treat degenerative diseases.Future work will focus on how to translate the concept of personalized medicine from bench work to clinical settings for targeted therapy to precisely and effectively use metformin in treating neurological diseases.
Author contributions:Manuscript writing:AL,CS,ML,LH,JW;literature search and collection:AL and JW;Figure and table creation:AL,CS,ML,JW;and manuscript design and conception:JW and LH.All authors approved the final version of the manuscript.
Conflicts of interest:The authors declare no conflicts of interest.
Data availability statement:All data relevant to the work are included in the article or uploaded as Additional files.
Open access statement:This is an open access journal,and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License,which allows others to remix,tweak,and build upon the work non-commercially,as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:Adriana Fernanda K Vizuete,Federal University of Rio Grande do Sul,Brazil.
Additional files:
Additional file 1:Open peer review report 1.
Additional Table 1:The positive effect of metformin on neurological diseases in humans and in vivo.
Additional Table 2:The moderate effect of metformin on neurological diseases in humans and in vivo.
Additional Table 3:The negative effect of metformin on neurological diseases in humans and in vivo.

- 中国神经再生研究(英文版)的其它文章
- From the dust: extracellular vesicles as regulators of development and neuroregeneration
- Targeting epidermal growth factor receptor signaling to facilitate cortical injury repair?
- Beyond functional MRI signals:molecular and cellular modifiers of the functional connectome and cognition
- Alpha7 nicotinic receptors as potential theranostic targets for experimental stroke
- Targeting autophagy by polyphenols to prevent glycative stress-toxicity in the brain
- Does photobiomodulation require glucose to work effectively?