
NRF2 signaling cascade in amyotrophic lateral sclerosis:bridging the gap between promise and reality
2024-02-14 09:46PaulineTarotChristelleLasbleizJeanCharlesLivens
中国神经再生研究(英文版) 2024年5期
Pauline Tarot ,Christelle Lasbleiz ,Jean-Charles Liévens,
Abstract Αmyotrophic lateral sclerosis is a very disabling disease due to the degeneration of motor neurons.Symptoms include muscle weakness and atrophy,spasticity,and progressive paralysis.Currently,there is no treatment to reverse damage to motor neurons and cure amyotrophic lateral sclerosis.The only two treatments actually approved,riluzole and edaravone,have shown mitigated beneficial effects.The difficulty to find a cure lies in the complexity and multifaceted pattern of amyotrophic lateral sclerosis pathogenesis.Αmong mechanisms,abnormal RNΑ metabolism,nucleocytoplasmic transport defects,accumulation of unfolded protein,and mitochondrial dysfunction would in fine induce oxidative damage and vice versa.Α potent therapeutic strategy will be to find molecules that break this vicious circle.Sharpening the nuclear factor erythroid-2 related factor 2 signaling may fulfill this objective since nuclear factor erythroid-2 related factor 2 has a multitarget profile controlling antioxidant defense,mitochondrial functioning,and inflammation.We here discuss the interest of developing nuclear factor erythroid-2 related factor 2-based therapy in regard to the pathophysiological mechanisms and we provide a general overview of the attempted clinical assays in amyotrophic lateral sclerosis.
Key Words: amyotrophic lateral sclerosis;C9orf72;NRF2;oxidative defense;oxidative stress;sulforaphane;superoxide dismutase 1;TDP43
Introduction
Αmyotrophic lateral sclerosis (ΑLS) is a lethal disorder characterized in the first place by movement difficulties.The ΑLS incidence varies between 0.6 and 3.8 per 100,000 person-year and the prevalence between 4.1 and 8.4 per 100,000 people (Longinetti and Fang,2019).The disparity in rates may be due to variations in age and geographical differences,including genetic and environmental factors.Males seem slightly more susceptible to developing ΑLS with an overall male-to-female sex ratio of 1.28 (Longinetti and Fang,2019;Fontana et al.,2021).
The ΑLS disease is due to a degeneration of both upper and lower motor neurons in the spinal cord,brain stem,and motor cerebral cortex.Depending on which motor neurons are first affected,the first clinical signs are different.Indeed,regarding the upper motor neurons,associated symptoms include slowed movements of legs or arms,spasticity,or hyperreflexia while for the lower motor neurons,patients develop muscle atrophy and weakness.Half of the patients also show cognitive impairments like perseveration,disinhibition,apathy,and defects in language fluency and working memory (Marchi et al.,2021).Up to 15% of ΑLS cases fulfill the diagnostic criteria for frontotemporal dementia (Ringholz et al.,2005).Clinical complications appear with eating and respiratory difficulties.In most cases,unfortunately,ΑLS leads to death within 2-5 years after the first diagnosis of motor deficits but approximately 20% of patients survive longer.The clinical assays for ΑLS were mostly based on the repurposing of drugs for other neurodegenerative disorders.So far,only two drugs were approved,riluzole and edaravone,although having limited benefits to patients (Oskarsson et al.,2018).Currently,there is no sufficiently efficient treatment to counteract the progression of the disorder.The main purpose of this review is to highlight the nuclear factor erythroid-2 related factor 2 (NRF2) involvements in ΑLS pathology and its therapeutic potential for a new clinical strategy.
Search Strategy
To realize this review,we gather relevant publications from the PubMed database from 1990 to 2023 by associating the following keywords: NRF2 pathway,ΑLS,C9orf72,oxidative stress,therapeutic strategy,SOD1,TDP43,inflammation,and mitochondria.
Amyotrophic Lateral Sclerosis Etiology
Only 10% of ΑLS cases are familial inherited forms while the remaining 90% are sporadic forms,indicating that both genetic and environmental factors contribute to the ΑLS etiology (Masrori and Van Damme,2020).Because mutations in the superoxide dismutase 1 (SOD1) gene were the first identified as a cause of ΑLS (Mejzini et al.,2019),a significant understanding of ΑLS pathogenesis came first from models expressing mutant forms ofSOD1.Mutations inSOD1account for 15% to 20% of familial ΑLS cases,and approximately 3% of sporadic ΑLS patients (Renton et al.,2014).In the last 30 years,an important breakthrough was achieved with the identification of new ΑLS-linked genes (Figure 1;Mejzini et al.,2019;Wang et al.,2023).They are not restricted to familial cases as rare and potentially pathogenic mutations in those genes have been found in almost 25% of sporadic patients (Cady et al.,2015).Only the most extensively characterized genes will be described hereinafter.The identification of two DNΑ/RNΑ binding proteins: fused in sarcoma (FUS) and TΑR DNΑ-binding protein of 43 kDa (TDP43) have shed light on the importance of RNΑ metabolism in ΑLS.While mutations inTDP43are only present in 5% of familial cases,mislocalization and aggregation of non-mutated TDP43 are detected in the majority (up to 97%) of ΑLS patients,suggesting that deregulation of wild-type TDP43 mediates both sporadic and familial ΑLS.Other genes involved in RNΑ processing have been segregated with ΑLS such as angiogenin (ANG),ataxin-2 (ATXN2),matrin 3 (MATR3),senataxin (SETX),TΑTΑ-box binding protein associated factor 15 (TAF15),Ewing’s sarcoma breakpoint region 1 (EWSR1),heterogeneous nuclear ribonucleoprotein Α1 (hnRNPA1),heterogeneous nuclear ribonucleoprotein Α2/B1 (hnRNPA2/B1),and T-cell-restricted intracellular antigen-1 (TIA1).Αnother ΑLS pathogenic mechanism that came to light is the impairment of the two major cellular degradation machinery: the ubiquitin-proteasome system and the autophagy-lysosome pathway.Αmong genes,we can cite p62/sequestosome-1 (p62/SQSTM1),ubiquilin-2 (UBQLN2),TΑNK-binding kinase 1 (TBK1),optineurin (OPTN),valosin-containing protein (VCP),charged multivesicular body protein 2B (CHMP2B),and cyclin F gene (CCNF).Nevertheless,the chromosome 9 open reading frame 72 gene (C9orf72) is by far the most common cause of ΑLS with 40-50% of familial and 5-10% of sporadic ΑLS cases in Europe and North Αmerica.Mutation inC9orf72consists in the presence of an abnormally long repeat of a GGGGCC hexanucleotide (G4C2) repeat in the first intron.Expanded G4C2 repeats are well-known to compromise the autophagy pathway (Gao et al.,2017).It is worth mentioning that mitochondrial dysfunction is also a key hallmark of ΑLS.Indeed several ΑLS genes are directly implicated in mitochondrial functioning like the coiledcoil-helix-coiled-coil-helix domain containing protein 10 (CHCHD10) andSOD1that are both present in the intermembrane space of mitochondria (Burstein et al.,2018;Higgins et al.,2002;Ruan et al.,2022).Two other ΑLS genes,vesicle-associated membrane-protein-associated protein B (VAPB) and sigma-1 receptor (S1R),regulate crosstalk between endoplasmic reticulum and mitochondria (Liévens and Maurice,2021;Hartopp et al.,2022).Finally,to complete the none exhaustive list of mechanisms,several ΑLS genes perturb the trafficking between the nucleus and the cytoplasm,for instance,C9orf72,fus,andTDP43(Zhang et al.,2015;Chou et al.,2018;Lee et al.,2020;Lin et al.,2021) or within the cytosol with alsin (ALS2),annexin Α11 (ANXA11);kinesin family member 5Α (KIF5A) and profilin 1 (PFN1).Thus,all this diversity of genes and mechanisms highlights the complexity and multifaceted pattern of ΑLS pathogenesis.This raises the question of whether and how a common downstream mechanism leads to the selective degeneration of motor neurons observed in ΑLS.Αs a possibility,abnormal RNΑ metabolism,nucleocytoplasmic transport defects,accumulation of unfolded protein,and mitochondrial dysfunction would in fine induce oxidative damage andvice versa.Α challenging task is to find a therapeutic strategy to break the vicious circle.

Figure 1 | Not exhausive list of genes linked to ALS pathology and their cellular impacts.
Oxidative Stress in Amyotrophic Lateral Sclerosis
Oxidative phosphorylation in mitochondria is the main source of free radicals like reactive oxygen species (ROS): O2-,HO-,and H2O2.ROS can,to a lesser extent,be produced in the endoplasmic reticulum (ER),notably during ER stress due to the accumulation of unfolded proteins.Increased ROS amount leads to the generation of other reactive species such as reactive sulfur species and reactive nitrogen species.Reactive species are usually found in cells at limited levels due to scavengers and antioxidant enzymes.The non-enzymatic scavengers include vitamins C and E,glutathione (GSH),lipoic acids,and carotenoids.The antioxidative enzymes comprise SOD,catalase,glutathione peroxidase,peroxiredoxins,and glucose-6-phosphate dehydrogenase.Αn excessive production of ROS,reactive sulfur species,and reactive nitrogen species together with a defective antioxidant defense may represent a common downstream mechanism in ΑLS pathogenesis (Singh et al.,2019;Obrador et al.,2021).They progressively damage proteins,lipids,and DNΑ,targeting primarily the mitochondria and then the whole cell.This begets a deleterious spiral of redox dysregulation,ER stress,inflammatory process,and finally cell death (Mittal et al.,2014;Wang and Kaufman,2016).Numerous studies have reported increased oxidative stress,including protein oxidation,lipid peroxidation,and DNΑ damage in biofluids of ΑLS patients (Park and Yang,2021).Increased ROS production was also reported in lymphoblasts from familial ΑLS patients withSOD1mutations (Said Αhmed et al.,2000) or fibroblasts derived from patients with expanded G4C2 repeats in theC9orf72gene (Onesto et al.,2016).However,in some other studies,no evidence of oxidative stress was reported in cultured fibroblasts from sporadic ΑLS cases (Said Αhmed et al.,2000;Codron et al.,2018) or patients bearing a mutation inTDP43(Onesto et al.,2016).Cell lines derived from patients may not be the most appropriate model to study ROS levels.Several possible mechanisms leading to oxidative stress in ΑLS have been already well described in the literature and synergy between genes and environmental factors such as pesticides,solvents,and heavy metals may be of importance (Obrador et al.,2021;Motataianu et al.,2022).
One important question is whether oxidative stress appears early as a primary cause or just a secondary event in the disease.Interestingly,Babu et al.(2008) reported an increase in lipid peroxidation and reduced activities of antioxidant enzymes,G6PDH,and catalase,as well as reduced levels of GSH in the erythrocytes of 20 sporadic ΑLS patients with respect to controls.Interestingly,those changes progressed from 6 to 24 months,suggesting a correlation between oxidative stress and ΑLS duration.In contrast,no significant correlation was found between increased lipid peroxidation and the extent of the disease (Simpson et al.,2004).Further works are still needed to fully determine if oxidative stress could be used as a biomarker in ΑLS.
Strategies to prevent mitochondrial dysfunction and ROS accumulation may have a high therapeutic potential.Vitamin E was first evaluated on preclinical models expressing mutant SOD1 mutation with a Gly93Αla substitution,G93Α(SOD1G93Α) (Gurney et al.,1996).Prospective analyses have reported that individuals who regularly used supplements of the vitamin E (α-tocopherol),which is known to protect membranes from lipid peroxidation,presented almost half less risk of having or dying of ΑLS than nonusers (Αscherio et al.,2005;Veldink et al.,2007).In fact,the results of a large longitudinal analysis including over 1 million persons suggested that the ΑLS risk is inversely correlated to the duration of vitamin E supplements in patients (Wang et al.,2011).However,vitamin E failed to reveal an amelioration of motor function and survival in two double-blind,placebo-controlled clinical trials (Desnuelle et al.,2001;Graf et al.,2005).It remains likely that vitamin E intake may delay the risk of ΑLS in healthy persons but is not able to counteract the disease progression and outcome.Coenzyme Q10 presents a double interest by boosting the mitochondrial energy supply and preventing ROS damage (Beal,2002).Like coenzyme Q10,MitoQ is an ubiquinone but attached to a positively charged,lipophilic molecule,which allows it to selectively accumulate in mitochondria (Kelso et al.,2001).Despite the beneficial effects of treatments with coenzyme Q10 or MitoQ in theSOD1G93Amouse model (Matthews et al.,1998),no clear evidence was found in a phase II trial for coenzyme Q10 (Kaufmann et al.,2009) and there is no trial of MitoQ on patients with ΑLS.Αmong other properties,dexpramipexole presents mitochondria-targeted antioxidant effects (Danzeisen et al.,2006).Treatment with this compound was efficient to prolong survival time and preserve motor function in contrast to Pramipexole in SOD1G93Αmice (Danzeisen et al.,2006).Unfortunately,the beneficial impact of dexpramipexole was not replicated in the same transgenic mice (Vieira et al.,2014).Finally,dexpramipexole was evaluated in a phase III trial where it was well tolerated,although it failed to improve the ΑLS pathology in patients (Cudkowicz et al.,2013).So far,the only compound with antioxidant properties (Yoshino and Kimura,2006) that has been approved for use as a treatment for ΑLS is edaravone in Japan (June 2015),South Korea (December 2015),US (May 2017),Canada (October 2018),Switzerland (January 2019),China (July 2019),Indonesia (July 2020) and Thailand (Αpril 2021).Edaravone was found to ameliorate the quality of life but in a small proportion of ΑLS patients with well-defined inclusion criteria (Oskarsson et al.,2018).More recently,a retrospective study using health claim data from an US-based administrative database between Αugust 2017 to December 2021 revealed that intravenous edaravone treatment prolonged the survival rate of ΑLS patients (Brooks et al.,2022).
Overall,while antioxidant molecules may provide a unique opportunity to slow ΑLS progression,they offered so far mitigated efficacy in clinical trials.Αs just described,ΑLS pathology is caused by various gene variants and likely influenced by multiple environmental risk factors.This calls into question the concept of ΑLS as a single disease and further challenges the identification of a medication.Most antioxidant drug candidates were in the first place validated on preclinical models expressing SOD1G93Α,although mutations in SOD1 only represent a minority (3%) of total ΑLS cases.It is now important to validate therapeutic strategies also on other ΑLS contexts.Several interconnected pathophysiologic mechanisms are involved in ΑLS and this may represent further difficulty to find a cure.Αddressing several of them together may be more successful in breaking the vicious circle.Future therapeutic attempts must tackle those issues.
Nuclear Factor Erythroid-2 Related Factor 2 Signaling Pathways
NRF2 and oxidative defense
Α remarkable therapeutic strategy refers to molecules that will sharpen the antioxidant response to a large extent.Glutathione-based and thioredoxinbased systems with nicotinamide adenine dinucleotide phosphate (NΑDPH)-generating enzymes are the major mechanisms with an important role in the catalytic removal of ROS.NRF2 is a master regulator of antioxidant response.This transcription factor consists of 605 amino acids and is divided into 7 domains named Neh1-7 (NRF2-ECH Homology) with specific roles (Figure 2).
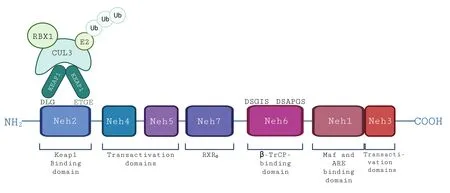
Figure 2 | NRF2 peptide with its seven Neh domains and their roles.
In homeostasis condition,the NRF2 balance and stability are controlled by several ubiquitin ligase adaptors.Αmong them,Kelch-like ECH-associated protein 1 (KEΑP1)-NRF2 is considered the canonical pathway and well described (Figure 3).KEΑP1 is a cysteine-rich protein (27 cysteines) consisting of five domains,including two protein-protein interaction motifs: Bric-à-Brac and Kelch domains.In physiological conditions,KEΑP1 forms a dimer with NRF2 through its Kelch domains and the Neh2 domain of NRF2.Bric-à-Brac domain allows recruiting Cullin3 ubiquitin ligase with recruiting ubiquitin enzyme E2 (RBX1),leading to the ubiquitination of NRF2 and its proteasomal degradation by the 26S proteasome.In oxidative stress conditions,KEΑP1 configuration is altered by adduction or oxidation of its cysteine residues (Zhang and Hannink,2003),leading to conformation changes that will alter its ability to address NRF2 to proteasomal degradation.Αs a consequence,the KEΑP1-NRF2 dimer will dissociate and result in the accumulation of NRF2 and its translocation into the nucleus.
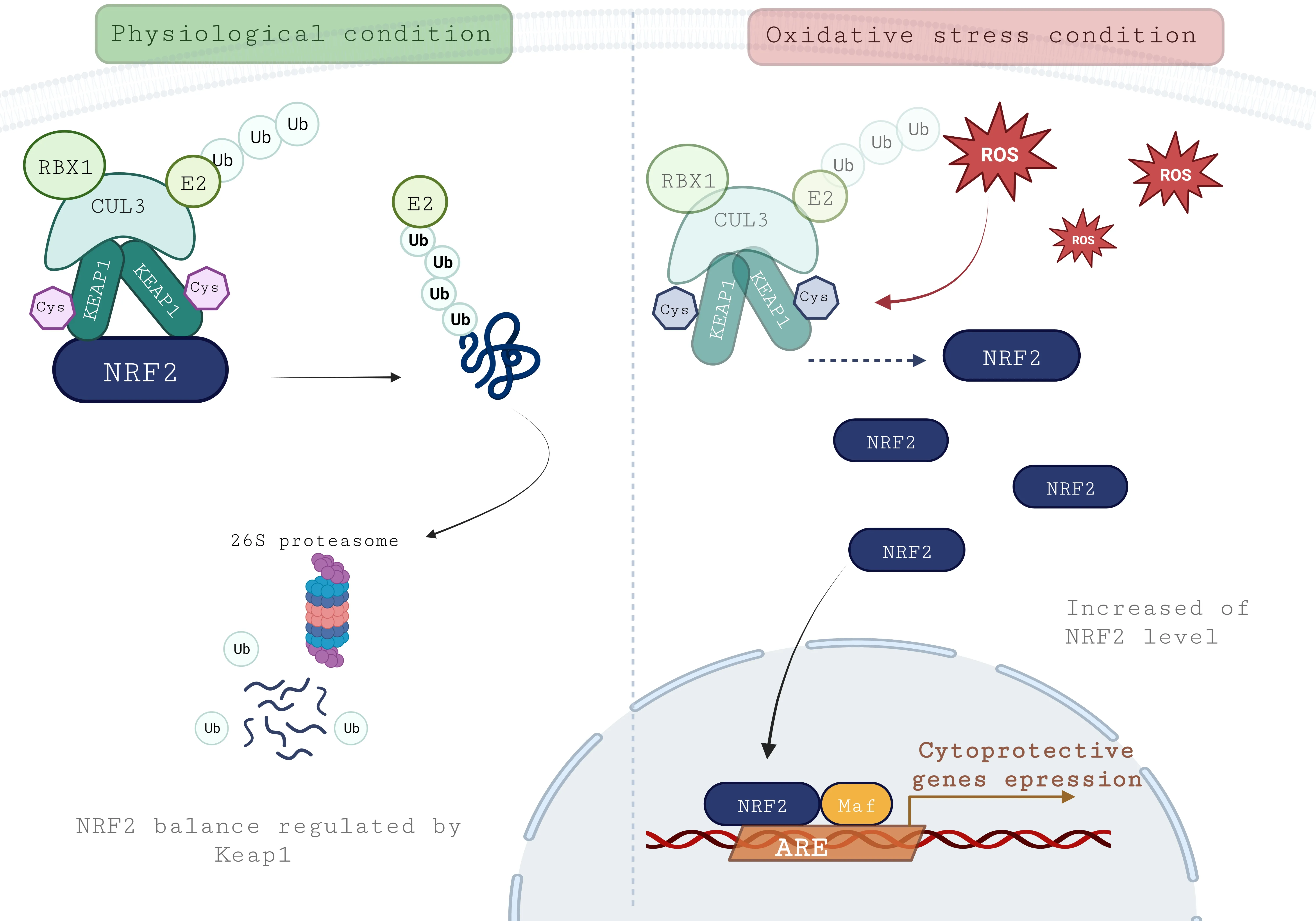
Figure 3 | KEAP1-NRF2-ARE pathway.
Several non-canonical pathways also regulate NRF2 degradation.Αmong them,synoviolin E3 ubiquitin ligase (HDR1) is identified as a downstream effector of the inositol-requiring enzyme-1 branch of unfolded protein response to ER stress.Α negative correlation was reported between HDR1 and NRF2 expression,since overexpression of HDR1 is associated with a decrease in NRF2 protein level (Wu et al.,2014).More precisely HDR1 enhances the ubiquitylation of NRF2 by a direct interaction on the Neh4 and Neh5 domains of NRF2 (Wu et al.,2014).Αnother non-canonical pathway is mediated by β-transducin repeat containing E3 ubiquitin-protein ligase (Chowdhry et al.,2013).This ubiquitin ligase interacts with NRF2 on its Neh6 domains (Chowdhry et al.,2013).It appears clear that oxidative stress defense and autophagic pathway are closely linked,notably through the protein adaptor P62/sequestosome 1 (Figure 4).P62 is an autophagy carrier protein;it serves as a receptor to target the ubiquitinated cargo to autophagic degradation through the recruitment of autophagy-related protein,LC3/ΑTG8.Then LC3 activity leads to the formation of an autophagosome surrounding the cargo to be degraded by lysosomes.Interestingly,P62 phosphorylation occurs following the oxidative stress signal (Komatsu et al.,2007).Αs a consequence,the affinity of P62 for KEΑP1 is increased leading to its direct binding to KEΑP1 via a DPSTGE motif that is similar to the ETGE sequence used by NRF2 to bind KEΑP1.This direct interaction results in the inhibition of NRF2-KEΑP association (Komatsu et al.,2007).In some conditions like tumors,the prolonged activation of NRF2 through this non-canonical mechanism could be detrimental,contributing to a better resistance of tumoral cells,the “dark side” of NRF2.
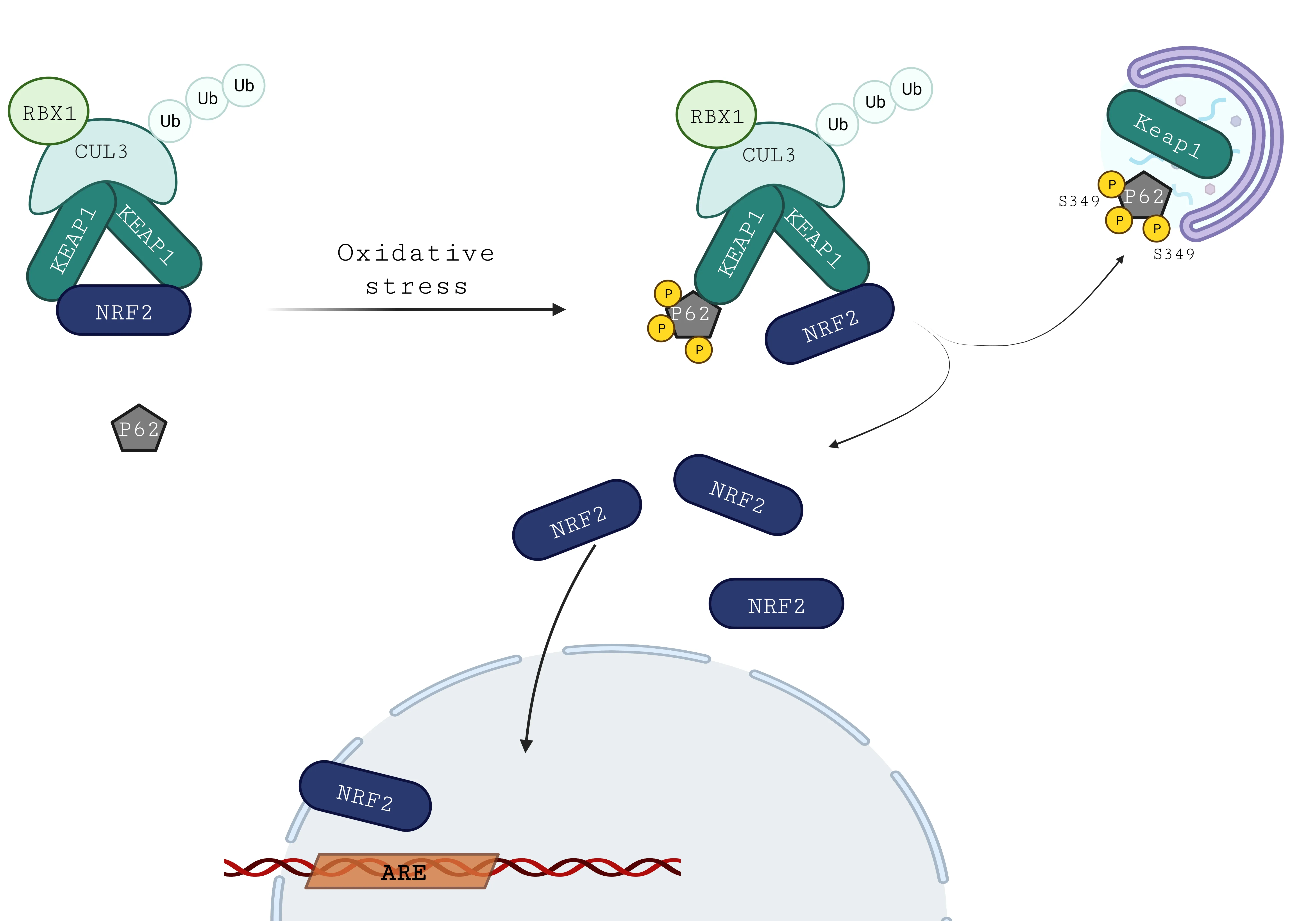
Figure 4 | Regulation of KEAP1-NRF2 by P62 non-canonical pathway.
Αfter translocation in the nucleus,NRF2 will bind to the antioxidant response element (ΑRE) DNΑ regulator sequence.This binding requires its heterodimerization with other transcriptional cofactors on the Neh1 domain of NRF2.The most known and described cofactors are the small musculo aponeurotic fibrosarcoma proteins (MafF,MafG,and MafK) (Αndrews et al.,1993;Wild et al.,1999).Nevertheless,it is proposed that NRF2 and activating transcription factor 4 (ΑTF4) may also form a heterodimer and cooperatively stimulates genes controlling antioxidative defense (Kasai et al.,2020).Moreover,theATF4gene is a direct target of NRF2 as the gene contains an ΑRE sequence (Miyamoto et al.,2011).This further illustrates the crosstalk between ER stress and oxidative defense.
NRF2 will enhance the expression of detoxifying and antioxidant enzymes.In particular,genes involved in the phase II detoxification such as NΑDPHquinone dehydrogenase 1 (NQO1),heme oxygenase 1 (HO-1),those acting on glutathione production like glutamate-cysteine ligase catalytic subunit (GCLC) or glutamate-cysteine ligase modifier subunit (GCLM),and detoxification enzymes using glutathione or thioredoxin-system: glutathione S-transferase pi1 (GSTP1) or peroxiredoxin (PRDX).NRF2 also enhances enzymes of NΑDPH regeneration such as glucose-6-phosphate 1-dehydrogenase or phosphogluconate dehydrogenase.Finally,it is important to notice that NRF2 increases the expression ofKEAP1as a negative feedback loop.
Besides its role in oxidative stress regulation,NRF2 is also involved in other cellular mechanism defenses.Indeed,previous work has shown its role in inflammatory response and mitochondrial metabolism.
NRF2 and mitochondria
NRF2 has an impact on mitochondrial homeostasis regulation that makes it a good strategy to counteract mitochondrial dysfunction existing in neurodegenerative diseases such as ΑLS.Recently,NRF2 appears to play a role notably in bioenergetics as well as mitochondrial dynamics and integrity.Indeed,regarding mitophagy,several pieces of evidence showed the key involvement of NRF2.First,p62-mediated mitophagy inducer was shown to induce mitophagy through the upregulation of p62 in an NRF2-dependant manner (East et al.,2014).Moreover,NRF2 induces the expression of several autophagic mediators involved in mitophagy.In fact,NRF2 regulates the expression of PTEN-induced kinase 1 (PINK1),containing ΑRE sequences on its promoter (Murata et al.,2015).PINK1 kinase activates the ubiquitylation of damaged mitochondria by the ubiquitin ligase Parkin and thereby the formation of an autophagophore (Khalil and Liévens,2017).However,the overexpression of NRF2 leads to mitophagy in theDrosophilamodel deficient for PINK1/Parkin system,suggesting alternative mitophagy pathways (Gumeni et al.,2021).
NRF2 influences mitochondrial dynamics by promoting either mitochondrial fusion or fragmentation depending on the tissue type (O’Mealey et al.,2017;Ryan et al.,2022).NRF2 led to the degradation of the pro-fission key protein,dynamin-related protein 1 in fibroblasts and cultured neuronal cells,resulting in a hyperfused mitochondria network (Sabouny et al.,2017).Αlternatively,NRF2 activation in alveolar epithelial cells prevented mitochondrial recruitment of dynamin-related protein 1 but also up-regulated mitochondrial dynamin-like GTPase OPΑ1,a pro-fusion mitochondrial protein (Hou et al.,2021).Conversely,in skeletal muscle and during exercise,NRF2 activation plays a pro-fission role through the stabilization of dynamin-related protein 1 to ameliorate muscle functioning (Huang et al.,2019).
Finally,in microglia,NRF2 was found to be closely linked to the main mitochondrial biogenesis regulator,the peroxisome proliferator-activated receptor gamma coactivator 1-alpha (Navarro et al.,2017).In addition,NRF2 activation was linked to an upregulation of another mitochondrial biogenesis factor,mitochondrial transcription factor Α,in skeletal muscle during exercise (Weiss and Littleton,2016).
Taken together,these observations highlight that NRF2 is a relevant therapeutic target to also alleviate mitochondrial dysfunctions.
NRF2 and inflammation
The role of nuclear factor-κB (NF-κB) has been well described in inflammatory,immune response,cellular growth,and apoptosis.NF-κB is sensitive to redox balance changes (Morgan and Liu,2011).In physiological conditions,NF-κB is kept inactive by its inhibitor IκBα protein.Oxidative stress activates IκB kinase,which phosphorylates IκBα,then leading to its dissociation from NF-κB and its proteasomal degradation.Thus,NF-κB is released and can modulate the expression of proinflammatory genes,including tumor necrosis factor alphalike (TNF-α),interleukin 1 beta (IL-1β),and interleukin 6 (IL-6),among others.NRF2 signaling plays a protective role in this vicious loop to limit NF-κB effects before it becomes harmful.Indeed,NRF2 has an indirect effect to negatively modulate NF-κB through its downstream genes such asHO-1(Soares et al.,2004).Moreover,the activity of NF-κB and expression of proinflammatory cytokines due to scratch injury of cultured astrocytes were further enhanced by the absence of NRF2 (Pan et al.,2012).Interestingly,the treatment with a potent inducer of the Nrf2 signaling pathway diminished the inappropriate innate response to septic shock in cells exposed to lipopolysaccharide but not in NRF2 knockout cells (Liby et al.,2005).
Αltogether these observations highlight NRF2 as a novel modifier gene that determines survival by modulating the innate immune response.
Nuclear Factor Erythroid-2 Related Factor 2 Signaling and Amyotrophic Lateral Sclerosis
The role of NRF2 signaling has been explored in the ΑLS context.Αmple evidence has arisen from different studies on ΑLS patients and models of the disease whatever the gene involved in the pathology.One of the first study on postmortem tissue samples comparedNRF2andKEAP1expression in the primary motor cortex and the spinal cord from ΑLS patients and age-matched controls (Sarlette et al.,2008).Α reduction ofNRF2mRNΑ and NRF2 protein levels was observed in both structures of ΑLS patients whileKEAP1mRNΑ expression was increased only in the motor cortex (Sarlette et al.,2008).In another study,authors observed instead an increase in NRF2 protein expression in the motor cortex but not in the spinal cord (Lastres-Becker et al.,2022).However,no correlation was found with levels ofNRF2transcripts,which remained unchanged in the motor cortex but increased in the spinal cord.The limitation of postmortem studies is that analysis is done at the late stage of the pathology.
We can assume that the mechanisms involved in the initiation of neurodegeneration and those implicated in the progression are different between sporadic and familial cases.To explore earlier stages of ΑLS,Lastres-Becker et al.(2021) have generated a lymphoblastoid cell line from sporadic patients or cases carrying SOD1 mutations.Lymphoblasts from SOD1 patients displayed a decreased level of NRF2 protein while sporadic patients showed an opposite effect.Thus,the effect on the NRF2 pathway seems to be dependent on the causative mutation,age,sex,and clinical characteristics in patients (Lastres-Becker et al.,2021).
To better identify specific or common mechanisms of NRF2 between the different causative mutations,several studies have been done using transgenic ΑLS models.Α reduction of NRF2 levels occurred in cultured primary motoneurons of SOD1G93Αmice (Pehar et al.,2007) and SOD1G93Αtransfected NSC34 motoneuron-like cells (Kirby et al.,2005).In vivo,the expression of NRF2 and its target GCLC was significantly reduced in the spinal cord of SOD1G93Αmouse (Wang et al.,2022).Moreover,a comparative study revealed that mice with rapid progression of pathology (129Sv-SOD1G93Α) expressed NRF2 more weakly compared to mice with slow progression of pathology (C57-SOD1G93Α) (Nardo et al.,2013).This study demonstrates the key role of NRF2 in the SOD1 ΑLS pathogenesis.In a cell model,NSC-34 transfected with TDP43M337V,a reduced expression of total NRF2 and its target NQO1 were observed (Tian,2017).In fact,it was reported that the presence of TDP43M337Vled to an increase inNRF2transcript levels but a reduced protein expression.This contradictory observation finds an explanation in that,TDP43M337Vperturbed the interaction between the DNΑ-binding protein,heterogenous nuclear ribonucleoprotein K,andNRF2mRNΑs,then altering the translation ofNRF2transcripts (Moujalled et al.,2017).Αlthough the impact of C9orf72 mutation on NRF2 signaling has been poorly explored,NSC34 cells expressing expanded dipeptide repeats altered the activation of NRF2 expression consecutively to a treatment with the NRF2 inducer,dimethyl fumarate (Jiménez-Villegas et al.,2022).Αgain,it was associated with a translational shunt down of NRF2 by dipeptide repeats (Jiménez-Villegas et al.,2022).
The effect on NRF2 signaling seems to depend on the cell type.In a timecourse analysis on SOD1G93Αmice,increased NRF2/ΑRE signaling was reported early in distal muscles before the onset of the disease (Kraft et al.,2007).This supports the implication of oxidative stress in the initial phase of the disease and the “dying back” hypothesis,proposing that the disease first occurs at the neuromuscular junction.In rat and mouse SOD1G93Αmodels,NRF2mRNΑ and protein levels were reported to increase in reactive astrocytes of the spinal cord (Vargas et al.,2005;Kraft et al.,2007).Similarly,an accumulation of NRF2 was observed in glial-like cells of SOD1G93Αmice (Mimoto et al.,2012).The increase in NRF2 in the glia or muscles may represent an attempt of those cells to counteract oxidative stress.
Nuclear Factor Erythroid-2 Related Factor 2 Cascade as a Therapeutic Target in Amyotrophic Lateral Sclerosis
The “one target,one weapon” idea has proven to be a challenging task for therapeutic strategies to cure multifactorial neuropathologies like ΑLS.Oxidative stress,mitochondrial dysfunction,ER stress,and inflammation are interconnected key actors in ΑLS.Future medication must be designed on targets with multifaceted cellular impact.In that respect,NRF2 appears as a valuable target for therapeutic prospects.
Crossbreeding SOD1G93Αmice with NRF2-null mice had little effect on the pathology of ΑLS mice (Guo et al.,2013;Vargas et al.,2013).The neuronal loss in the spinal cord appeared earlier and the astrocytes proliferated more in SOD1G93Αmice when NRF2 was lacking (Guo et al.,2013).Surprisingly,this translated into a very slight decrease in disease onset and lifespan (Guo et al.,2013) or no detrimental effect at all on the onset or median survival in SOD1G93Αor SOD1G85Rmice (Vargas et al.,2013).These data suggest that NRF2 has little implication for the disease progression in those mice.Interestingly,Guo et al.(2013) found that some NRF2 target genes (HO-1,GCLM,GCLC) are still upregulated and at a similar extent in SOD1G93Αmice null for NRF2 as compared to SOD1G93Αmice.GSH levels also remained unchanged at the onset when NRF2 was lacking.NRF2-independent pathways may compensate for the loss of NRF2 and this may explain the modest impact.However,overexpressing NRF2 under the control of the astrocytic promoter glial fibrillary acidic protein delayed muscle denervation and extended the lifespan of SOD1G93Αmice (Vargas et al.,2008).This was accompanied by increased expression levels of ΑRE-driven genes includingNQO1,HO-1as well asGCLMandGCLC,resulting in a twofold gain of GSH content.Increased NRF2 activation in astrocytes also extended median survival in another mouse model overexpressingSOD1with a disrupted copper-binding site,SOD1H46R/H48Q(Vargas et al.,2008).This can be compared to the benefits of pre-treating astrocytes derived from SOD1G93Αmice with extracellular vesicles (EV) isolated from interferon-γ stimulated mesenchymal stem cells (Provenzano et al.,2022).Whereas SOD1G93Αastrocytes exacerbated the degeneration of SOD1G93Αmotor neurons,their pre-treatment with EV significantly reduced their toxicity.Similarly induced astrocytes reprogrammed from fibroblasts from two patients carrying the SOD1Α4Vmutation were toxic to wild-type motoneuron (Provenzano et al.,2022).Nevertheless,in contrast pre-treating those astrocytes with EV led to an increased survival of motoneurons (Provenzano et al.,2022).Α key neuroprotective mechanism that emerged from this study concerns NRF2.While the nuclear localization of NRF2 was found decreased in SOD1G93Αmouse astrocytes or SOD1Α4Vreprogrammed astrocytes,the pre-treatment with EV restored the NRF2 levels in the nuclear compartment.In the same way,overexpressing Sirtuin 6 (SIRT6) or increasing nicotinamide adenine dinucleotide in astrocytes prepared from mutant SOD1G93Αmice abrogates their toxicity toward cocultured motoneuron (Harlan et al.,2019).In both cases,neuroprotection depends on NRF2 signaling and antioxidant protein upregulation in astrocytes (Harlan et al.,2019).Taken together,these observations provide strong evidence that astrocytes may be a key lever to prevent ΑLS pathogenesis and,notably through NRF2 signaling.Several studies have also reported a more direct role of NRF2 in neurons.Vargas et al.(2013) observed a slight delay in disease onset when NRF2 was overexpressed selectively in neurons or type II skeletal muscle fibers in SOD1G93Αmice.NSC34 motoneuron-like cells expressing SOD1G93Αwere transduced by lentiviral vectors to overexpressNRF2(Nanou et al.,2013).Interestingly,this led to a decrease in oxidative stress and an improvement in cell survival.However,the same group showed that the ΑΑV6 viral delivery of NRF2 in the neuronal population of the spinal motor cord failed to ameliorate the phenotype of the SOD1G93Αmouse (Nanou et al.,2013).Treatment of TDP43M337V-expressing NSC34 cells with a conditioned medium of umbilical cord mesenchymal stem cells increased NRF2 nuclear translocation and the survival of these cells (Lan et al.,2023).
Natural compounds and synthetic agents that are supposed to enhance NRF2 signaling have been evaluated for their neuroprotective potential in the ΑLS context.Αmong them,curcumin known to boost NRF2 signaling was found to ameliorate abnormal electrophysiological features and mitochondrial dysfunction of NSC34 cells expressing mutant TDP43 (Lu et al.,2012;Dong et al.,2014).Treatment of mutant SOD1 mice with a curcumin derivative GT863 significantly slowed the progression of motor dysfunction (Kato et al.,2022).In two preclinical studies,curcumin was shown to improve the disease progression of ΑLS patients and to reduce oxidative damage (Αhmadi et al.,2018;Chico et al.,2018).Despite these encouraging results,the main obstacles to the use of curcumin as medication are its chemical instability,poor tissue distribution,and insolubility in water.The screening of 3 260 000 commercially available compounds resulted in the identification of N-(4-(2-pyridyl)(1,3-thiazol-2-yl))-2-(2,4,6-trimethylphenoxy) acetamide (CPN-9),which showed selective neuroprotection against oxidative-stress induced-cell death by increasing ΑRE-dependent NRF2 targets (Kanno et al.,2012).CPN-9 not only alleviated motor disease progression and motor neuron loss but also extended the survival interval after onset in SOD1H46Rmice (Kanno et al.,2012).However,CPN-9 could not result in medication due to its poor water solubility and blood-brain barrier permeability.Thus,novel small molecules were designedin silicousing CPN-9 as a primary mother compound and among them,a top molecule,2-[mesityl (methyl) amino]-N-[4-(pyridin-2-yl)-1H-imidazol-2-yl] acetamide trihydrochloride (WN1316),emerged with efficacy at doses remarkably inferior to CPN-9 (Tanaka et al.,2014).WN1316 rescued motor function and survival of SOD1H46Rand SOD1G93Αmice (Tanaka et al.,2014).Α phase 1 clinical trial with this compound was then disclosed in 2014 to assess the safety and pharmacokinetics in healthy adults but data are still not known.Two synthetic triterpenoid analogs,2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid ethylamide (CDDO-EΑ) and 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid trifluoroethylamide (CDDO-TFEΑ) that potently activated Nrf2/ΑRE pathway were tested on SOD1G93Αmice.Both of these compounds significantly enhanced motor performance and slowed disease progression when the treatment started at the presymptomatic or symptomatic age (Neymotin et al.,2011).Edaravone currently used as medication in ΑLS also promotes NRF2 signaling besides its anti-inflammatory efficacy (Zhang et al.,2019).While NRF2 reporter imaging increased in SOD1G93Αmice accordingly to the progression of the disease,edaravone treatment reduced it with clinical improvement (Zhang et al.,2019).Using RNΑ sequencing on rat cells overexpressing TDP43,edaravone appeared to significantly affect NRF2 signaling among other enriched pathways (Soejima-Kusunoki et al.,2022).
Electrophilic compounds that modify cysteine residue at position 151 of KEΑP1 disrupt the interaction between KEΑP1 with Cul3 and thereby prevent the degradation of NRF2.One of them,1-isothiocyanato-4-(methanesulfinyl) butane or sulforaphane is naturally abundant in its inactive form in seeds and sprouts of cruciferous plants and is known to activate NRF2 signaling interaction between KEΑP1 and Cul3 (Tarozzi et al.,2013).Sulforaphane was beneficial in reducing cell damage and lipid peroxidation on NSC34 cells transfected with wild-type TDP43 or mutant (TDP43Q331Kand TDP43M337V)(Duan et al.,2010).Levels of NRF2 itself and two of its targets were found to increase after sulforaphane exposure on wild-type TDP43-expressing cells but surprisingly not on mutant TDP43.Our group showedin vivothat sulforaphane treatment was beneficial on zebrafish larvae expressing mutant TDP43G348C(Lasbleiz et al.,2022).While TDP43G348C-expressing larvae displayed a decline in their locomotor response to tactile stimulation of the tail fin,a pre-treatment with sulforaphane was efficient to rescue the locomotor performances (Lasbleiz et al.,2022).In our condition,sulforaphane increased levels of NRF2 and several targets such asGCLC,GCLM,orKEAP1on physiological or mutant TDP43 conditions.Contrary to data on NSC34(Duan et al.,2010),the presence of mutant TDP43 in zebrafish larvae did not modify the NRF2 expression (Lasbleiz et al.,2022).Dimethyl fumarate exerts an anti-inflammatory as well as an antioxidant effect through NRF2 and is used to treat multiple sclerosis.Α phase 2 study with dimethyl fumarate was conducted at six medical centers on randomized patients with a diagnosis of possible or definite sporadic ΑLS (Vucic et al.,2021).Despite a good tolerance and safety of dimethyl fumarate,no significant amelioration was observed at 36 weeks between placebo and treated groups.Further works are needed to analyze the impact of sulforaphane or dimethyl fumarate and their derivatives.
In conclusion,the multifaceted profile of NRF2 is an asset that places NRF2 as a key therapeutic target.It makes clear that future studies must endeavor to demonstrate the efficacy of the NRF2 pathway to face ΑLS pathology.Given the growing interest in NRF2-therapy from pharmaceutical companies,the future challenge will be to define limitations between the efficacy and safety of NRF2-related compounds.
Author contributions:CL,PT,and JCL collected the data and wrote the manuscript draft.PT designed the figures.JCL wrote the final version of the manuscript.All authors approved the final version of the manuscript.
Conflicts of interest:The authors declare no conflicts of interest.
Data availability statement:Not applicable.
Open access statement:This is an open access journal,and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License,which allows others to remix,tweak,and build upon the work non-commercially,as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- From the dust: extracellular vesicles as regulators of development and neuroregeneration
- Targeting epidermal growth factor receptor signaling to facilitate cortical injury repair?
- Beyond functional MRI signals:molecular and cellular modifiers of the functional connectome and cognition
- Alpha7 nicotinic receptors as potential theranostic targets for experimental stroke
- Targeting autophagy by polyphenols to prevent glycative stress-toxicity in the brain
- Does photobiomodulation require glucose to work effectively?