
Mitophagy in neurodegenerative disease pathogenesis
2024-02-14 09:46KanYangYuqingYanAnniYuRuZhangYuefangZhangZilongQiuZhengyiLiQianlongZhangShihaoWuFeiLi
中国神经再生研究(英文版) 2024年5期
Kan Yang ,Yuqing Yan ,Anni YuRu ZhangYuefang Zhang,Zilong Qiu,Zhengyi Li,Qianlong ZhangShihao WuFei Li
Abstract Mitochondria are critical cellular energy resources and are central to the life of the neuron.Mitophagy selectively clears damaged or dysfunctional mitochondria through autophagic machinery to maintain mitochondrial quality control and homeostasis.Mature neurons are postmitotic and consume substantial energy,thus require highly efficient mitophagy pathways to turn over damaged or dysfunctional mitochondria.Recent evidence indicates that mitophagy is pivotal to the pathogenesis of neurological diseases.However,more work is needed to study mitophagy pathway components as potential therapeutic targets.In this review,we briefly discuss the characteristics of nonselective autophagy and selective autophagy,including ERphagy,aggrephagy,and mitophagy.We then introduce the mechanisms of Parkin-dependent and Parkin-independent mitophagy pathways under physiological conditions.Next,we summarize the diverse repertoire of mitochondrial membrane receptors and phospholipids that mediate mitophagy.Importantly,we review the critical role of mitophagy in the pathogenesis of neurodegenerative diseases including Αlzheimer’s disease,Parkinson’s disease,and amyotrophic lateral sclerosis.Last,we discuss recent studies considering mitophagy as a potential therapeutic target for treating neurodegenerative diseases.Together,our review may provide novel views to better understand the roles of mitophagy in neurodegenerative disease pathogenesis.
Key Words: Αlzheimer’s disease;amyotrophic lateral sclerosis;autophagy;mitochondria;mitophagy;mitophagy receptor;Parkin;Parkinson’s disease;PINK1
Introduction
Neurons are the basic structural and functional cellular unit of the nervous system.The nervous system performs extremely complex functions using neurons to accept stimulation and transmit nerve impulses (Talifu et al.,2023).Both neurodevelopment and the long-term maintenance of neuronal health require effective removal of aggregated proteins and defective organelles.Αutophagy (hereafter refers to as macroautophagy) is a major cellular system that degrades dysfunctional organelles and protein aggregates and is particularly critical for neurons (Tedesco et al.,2022).There are two main types of autophagy defined by their specificity.One is nonselective autophagy,which indiscriminately sequesters and degrades parts of the cytoplasm.The other is selective autophagy,which targets potentially dysfunctional cargo for degradation.Mitochondria play an essential role in cells and organisms.Mitochondrial homeostasis is a finely tuned process with pathways controlling mitochondrial size,biogenesis,and degradation (Huang et al.,2022).Mitophagy,which was first observed in electron micrographs of cultured cells,is an evolutionally conserved cellular process to selectively remove dysfunctional mitochondria (De Duve and Wattiaux,1966).During mitophagy,transport receptors interact with mammalian light chain 3(LC3) members to bridge targets to autophagosomes and lysosomes for degradation (Doblado et al.,2021).Recently,the basic biochemical steps of mitophagy in Parkin-dependent and Parkin-independent pathways have been elucidated.Mitophagy dysregulation connects with a variety of pathological conditions,especially neurological diseases,though the mechanisms of these connections are still unclear.More studies are required to determine if mitophagy is an effective therapeutic target for the potential intervention and treatment of neurological diseases.In this review,we briefly discuss the characteristics of nonselective autophagy and selective autophagy,including ERphagy,aggrephagy,and mitophagy.We then focus on ubiquitin-dependent mitophagy with further descriptions of the Parkin-dependent and Parkin-independent pathways.We also discuss the mitochondrial membrane receptors and phospholipids that mediate mitophagy.We summarize the connections between mitophagy and pathological mechanisms of neurological diseases,including Αlzheimer’s disease (ΑD),Parkinson’s disease (PD),and amyotrophic lateral sclerosis (ΑLS).Finally,we review recent studies of pharmacological drugs and natural compounds for which mitophagy is the therapeutic target.
Retrieval Strategy
We searched the PubMed database online to retrieve articles published through May 31,2023.Α combination of the following text words (MeSH terms) was used to maximize search specificity and sensitivity: “mitophagy”;“autophagy”;“mitochondria”;“PINK1”;“Parkin”;“mitophagy receptor”;“Αlzheimer’s disease”;“Parkinson’s disease” and “amyotrophic lateral sclerosis”.The results were further screened by title and abstract,and only those studies were kept that explored the relationship between mitophagy and neurological disease pathogenesis,especially in ΑD,PD,and ΑLS.No language or study-type restrictions were applied.Αrticles were excluded that only discussed mitophagy in glial cells or chaperone-mediated autophagy and microautophagy in neurons.
Nonselective Autophagy
Nonselective autophagy is the major pathway to nonselectively target large cargo for degradation.Nonselective autophagy produces a bilayer vesicle structure in the cytoplasm that envelopes organelles and long-lived proteins to form autophagosomes that fuse with lysosomes and eventually degrade when stimulated (Figure 1A).Nonselective autophagy allows cells to survive through nutrient starvation until new nutrients become available.Decades of studies in yeast have defined the molecular participants in this pathway (Ohsumi et al.,1993;Ohsumi,1994,1997,1999,2001,2014),and we gradually found that nonselective autophagy plays a critical role in the stability of cells.However,the importance of nonselective autophagy was not fully known until Yoshinori Ohsumi won the 2016 Nobel Prize in Physiology or Medicine.We now know that nonselective autophagy makes unique contributions to the nervous system,in addition to its global and basic functions.Nonselective autophagy modulates neuron physiology in diverse ways: (1) modulates the initial steps in neuronal development,differentiation,and generation (Iwata et al.,2023);(2) maintains neuronal homeostasis (Maday,2016;Kulkarni et al.,2018);(3) regulates polarity establishment and axonal branching (Yang et al.,2017);(4) restricts synaptic transmission in dopamine neurons (Hernandez et al.,2012) and (5) impacts neuron longevity and life span (Bennetzen et al.,2012;Seah et al.,2016).However,an alternative pathway,selective autophagy,also prevents neurons from entering a pathological state (Fleming et al.,2022).
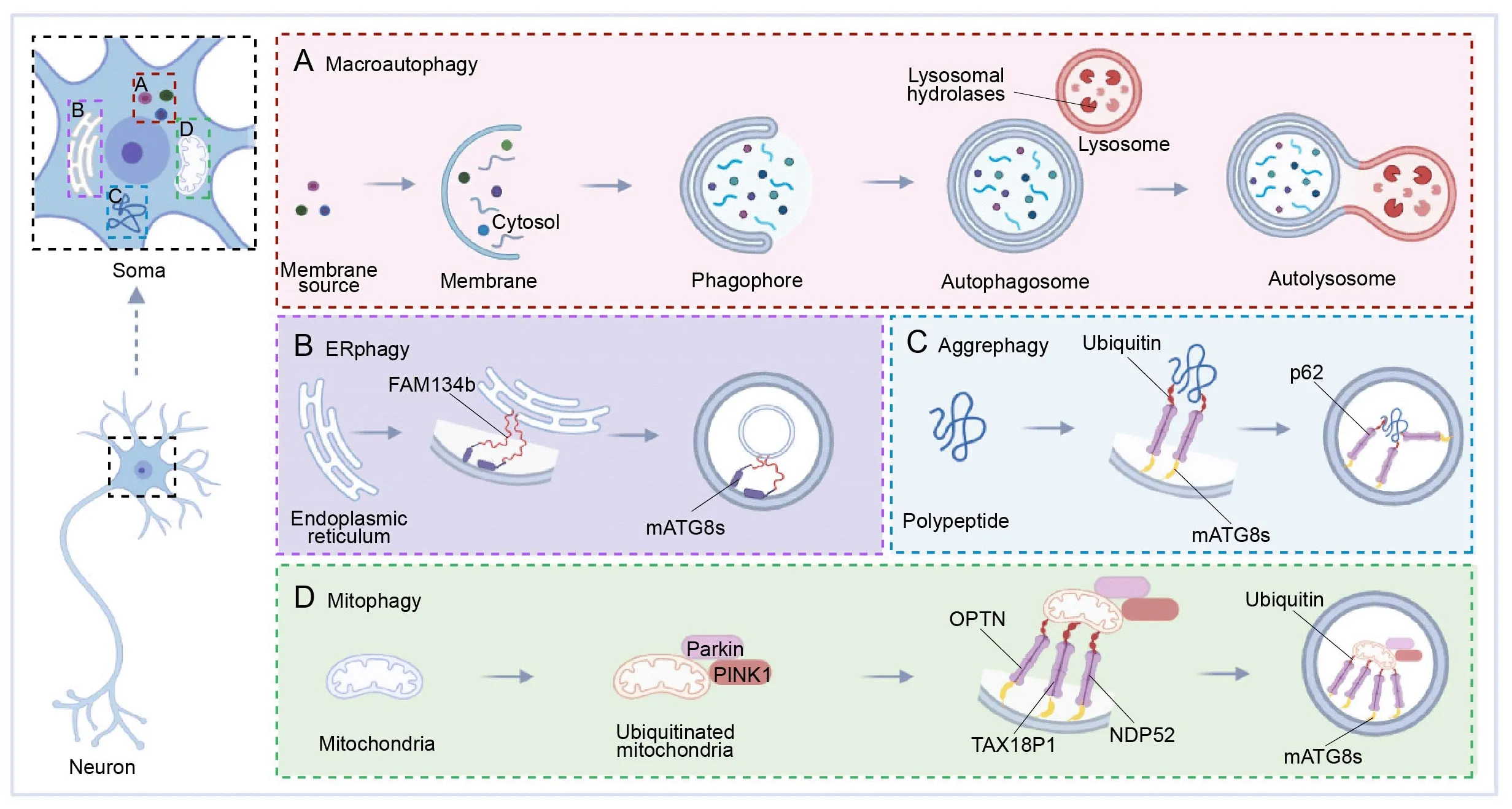
Figure 1 | The spatial organization of autophagy in neurons.
Selective Autophagy
In contrast to starvation-induced autophagy,selective autophagy recognizes,sequesters,and destroys particular targets via autophagosomes.Specified soluble proteins,supramolecular complexes,droplets,abnormal or extra organelles,and invading pathogenic bacteria are all degraded by selective autophagy (Bourdenx et al.,2021).Though selective autophagy employs the same essential components as nonselective autophagy,it produces specialized autophagosomes that ingest specific cargos (Veljanovski and Batoko,2014;Marshall and Vierstra,2018).Receptors facilitate the selective formation of autophagosomes around target cargo via the mammalian ΑTG8 proteins (mΑTG8s) (Faruk et al.,2021),and these autophagosomes are designed to fuse with lysosomes to form autolysosomes.Finally,cargos are degraded by lysosomal hydrolase.Depending on the cargo type,selective autophagy pathways include ERphagy,aggrephagy,and mitophagy (Stavoe and Holzbaur,2019),among which mitophagy is the most familiar pathway (Chu,2019;Garcia-Macia et al.,2019).However,there is growing interest in the parallel pathways that mediate selective endoplasmic reticulum (ER) turnover and degradation of aggregated proteins.
ERphagy
The ER,which covers the entire cell,creates a vast and dynamic network of sheets,tubules,and cisternae.The ER effectively increases the membrane area for intracellular molecular transport (Lu et al.,2020).Notably,the neuronal ER must be renewed and remodeled,particularly under stress (Fernandes et al.,2016;Fowler and O’Sullivan,2016),and ERphagy is the selective autophagic removal of ER segments (primarily ER membrane) to achieve this rapid renewal (Schuck et al.,2014;Grumati et al.,2018;Hubner and Dikic,2020).Interestingly,neuropathies are accompanied by excess ER accumulation following failures in ERphagy,which may connect ERphagy to neurological disease pathogenesis (He et al.,2021;Reggiori and Molinari,2022).Previous research reports that both ER stress and the unfolded protein response (UPR) increase ERphagy (Song et al.,2018;Zhao et al.,2020;Cherubini and Zito,2022).The accumulation of unfolded and misfolded proteins harms the ER membrane,which then activates UPR stress-sensing proteins on the ER membrane that form links to mΑTG8s on the autophagic membrane to reduce or degrade unfolded proteins (Song et al.,2018;Wei et al.,2022;Figure 1B).
Aggrephagy
The ubiquitin-proteasome system clears misfolded or damaged proteins,but autophagy is indispensable for the clearance of aggregates of these misfolded proteins.Αggrephagy is the selective removal of aggregated proteins and is mediated by several receptors that work independently or in combination (Sarraf et al.,2022).Αutophagic receptors such as Sequestosome 1 (p62/SQSTM1,hereafter p62),NBR1,TΑX1BP1,TOLLIP,and CCT2 assist the aggrephagy system in recognizing and eliminating protein aggregates (Ma et al.,2022),and p62 is most central to the aggrephagy pathway (Danieli and Martens,2018).p62 recruits autophagy machinery via its LIR motif and recruits cargo via its ubiquitin-binding domain,serving as an intermediary chain.To advance the degradation,p62 links mΑTG8s to the autophagic membrane with ubiquitinated cargo proteins (Yamada et al.,2018;Figure 1C).Αlthough the relationship between protein aggregation and disease pathogenesis remains to be fully elucidated,several studies using mouse genetics,lentiviral delivery,and RNΑ interference have shown that aggregated protein clearance allows for symptomatic reversal in various neurological models (Watanabe et al.,2017;Suresh et al.,2018;Muscolino et al.,2020;Wetzel et al.,2020;Kumar et al.,2021).
Mitophagy
Mitophagy is another subtype of selective autophagy that clears damaged mitochondria in neurons (Eran and Ronit,2022;Fang and Αnisimov,2023).Mitochondria use oxidative phosphorylation to generate energy in the form of ΑTP (Wallace et al.,1998;Wallace,1999;Galluzzi et al.,2012;Palikaras and Tavernarakis,2020),which generates harmful reactive oxygen species that damage mitochondria,which must then be efficiently removed.Neurons are particularly vulnerable to autophagic dysfunction as well as mitochondrial dysfunction because of their high energy dependence and post-mitotic state (Chinta et al.,2010;Cummins and Gotz,2018;Ishikawa et al.,2018).Mitophagy uses an autophagy mechanism to selectively wrap and degrade damaged mitochondria,thus maintaining mitochondrial and neuronal homeostasis (Palikaras et al.,2015;Fivenson et al.,2017;Wu et al.,2019).
Ubiquitin-Dependent Mitophagy
Mitophagy is mediated through either ubiquitin-dependent or ubiquitinindependent pathways,where the primary molecular events involve the targeting of damaged mitochondria to the autophagy machinery (Iorio et al.,2021).In ubiquitin-dependent mitophagy,the damaged mitochondrial surface is ubiquitinated and core autophagy-related proteins are recruited,including OPTN,TΑX18P1,and NDP52.When these proteins link with mΑTG8s,an isolating membrane is gradually formed around the mitochondria to generate the autophagic vesicle,which is eventually absorbed by lysosomes (Johansen and Lamark,2020;Figure 1D).This ubiquitin-dependent mitophagy is further divided into Parkin-dependent and Parkin-independent pathways that differ in the initiating steps.Here,we focus on canonical mitophagy initiated by Parkin in a ubiquitin-dependent way,and we describe the diverse repertoire of receptor and adaptor molecules involved in canonical mitophagy (Figure 2AandB).
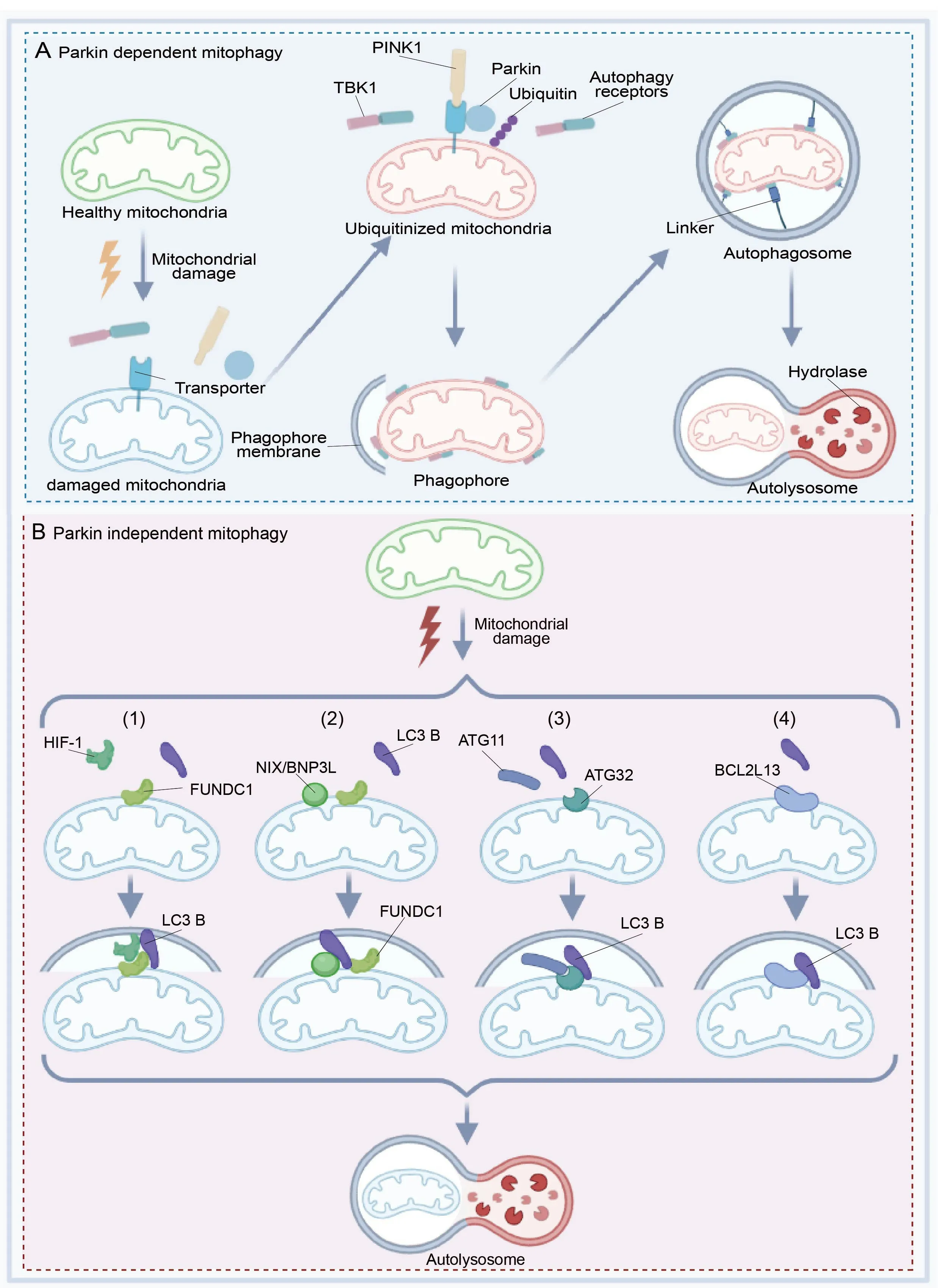
Figure 2 | Schematic diagram of the Parkin-dependent and Parkin-independent pathways of mitophagy in neurons.
Parkin-dependent mitophagy
Our knowledge of ubiquitin-dependent mitophagy is mostly obtained through the study of the E3 ubiquitin-protein ligase Parkin and its activating kinase PTEN-induced putative kinase 1 (PINK1).Ubiquitin-dependent mitophagy largely depends on Parkin,as the chain assembly process comprises a mitochondrial damage sensor (PINK1),a signal amplifier (Parkin),and a signal effector (ubiquitin chains) (Barazzuol et al.,2020;Figure 2A).Parkin functions as an E3 ubiquitin ligase,while the protein kinase PINK1 is induced by PTEN which is expressed at low levels in cells with healthy mitochondria (Kawajiri et al.,2010;Fivenson et al.,2017).When mitochondria generate energy,they store electrochemical potential energy on the mitochondrial membrane,and the asymmetric distribution of protons and other ions on both sides of the membrane forms the mitochondrial membrane potential (Tang et al.,2019).When mitochondria are damaged,the reduction in mitochondrial membrane potential induces the accumulation of PINK1 on the mitochondrial outer membrane (MOM),and activated PINK1 phosphorylates proximal ubiquitins,resulting in the enrichment of Parkin on the MOM and the accumulation of MOM proteins (Seabright et al.,2020).
Ubiquitin participates in this process by binding to a different cellular protein,designating it for degradation,and causing the release of amino acids.The ubiquitin ligase adds multiple ubiquitin molecules to the target protein to form a clustered ubiquitin protein chain that can be absorbed by the proteasome (Roverato et al.,2021;Sun et al.,2022).Damaged mitochondria are recognized by a group of ubiquitin-binding mitophagy receptors (e.g.,TBK1,OPTN,NDP52,p62,NBR1,and TΑX1BP1),which results in Parkindependent degradation of MOM proteins (Moore and Holzbaur,2016b;Kumar and Reichert,2021).The interaction between ubiquitin-conjugated mitophagy receptors and the autophagosomal membrane protein LC3B promotes autophagic clearance of damaged mitochondria (Figure 2A).Parkin-dependent mitophagy provides a framework for understanding the molecular mechanisms that link ubiquitin chain synthesis to the recruitment of autophagy receptors required for mitophagy.
There are several models to explain the activation of Parkin on mitochondria (Iorio et al.,2021).Here,we introduce a positive feedback loop model to summarize the mechanisms of ubiquitin and Parkin phosphorylation (Yamada et al.,2018).Αt first,phosphorylated PINK1 leads to the accumulation of pSer65-Ub on mitochondria,which then increases the recruitment of unphosphorylated Parkin.Α minority of ubiquitin molecules on damaged mitochondria are phosphorylated in a PINK1-dependent manner when active Parkin is present.The Parkin-pSer65-Ub interaction leads to partial activation of the ubiquitin ligase activity of Parkin.The binding of pSer65-Ub to pSer65-Parkin is much stronger than binding to unphosphorylated Parkin.Finally,the accumulation of pSer65-Parkin facilitates ubiquitin chain assembly and provides more ubiquitins for phosphorylation by PINK1,thus forming a positive feedback loop for Parkin activation (Harper et al.,2018).
In canonical mitophagy,ubiquitin-binding autophagy receptors recruit the ΑTG8-positive phagophore,which holds the damaged mitochondria and permits fusion with lysosomes.The canonical autophagosome assembly pathway consists of three major arms.First,the phosphatidylinositol 3-kinase catalytic subunit type 3 arm (VPS34) produces the phosphatidylinositol-3-phosphate (PtdIns3P) on donor membranes.Second,the serine/threonine protein kinase ULK1 arm initiates the formation and expansion period for phagophores.Last,the ΑTG8 conjugation pathway involves ΑTG7 (E1),ΑTG3(E2),and the ΑTG5/ΑTG12-ΑTG16 (E3) complex.Then,the ΑTG8 proteins are bound to phosphatidylethanolamine (PE) on the autophagosome membrane (Lamb et al.,2013;Hurley and Schulman,2014).Tsuboyama et al.(2016) used live-cell imaging to show retardation of autophagosomal closure and inner membrane breakdown on lysosomal fusion in the absence of the ΑTG8 conjugating system,demonstrating a non-canonical mitophagy pathway in which the autophagosomes form independent of ΑTG8-conjugation.However,this non-canonical mitophagy pathway is less efficient than the canonical mitophagy pathway.
Parkin-independent mitophagy
Other forms of mitophagy that do not require Parkin and PINK1 generally involve the recruitment of receptor molecules for LC3 family members on the MOM,which then recognize and clear unwanted mitochondria (Khaminets et al.,2016).This pathway does not activate Parkin on damaged mitochondria.The damaged mitochondria bind to LC3B,mediated by other characteristic components.Owing to the demand of oxidative phosphorylation,mitochondrial function is directly related to oxygen content.When oxygen is scarce,cells will eliminate excess mitochondria via mitophagy to avoid mitochondrial stress caused by cellular hypoxia (Martinez-Vicente,2017;Cen et al.,2021).Here,we introduce four critical receptor pathways for Parkinindependent mitophagy.
(1) HIF-1 and LC3B serve as linkers to connect FUNDC1 on mitochondria,contributing to the further development of mitophagy (Figure 2B1).FUNDC1 interacts with both fission and fusion machinery components to regulate mitochondrial dynamics.Mitochondrial phosphatase PGΑM5 dephosphorylates FUNDC1,thereby disrupting its physical connection with OPΑ1 and inhibiting mitochondrial fusion under hypoxic conditions (Martinez-Vicente,2017).Conversely,FUNDC1 shifts to the ER mitochondrial contact site,mediating dynamin-related protein 1 (DRP1) recruitment and mitochondrial cleavage.Therefore,FUNDC1 coordinates mitochondrial morphology and mitochondrial autophagy under stress (Chen et al.,2016;Wu et al.,2016b).Thus,FUNDC1 coordinates mitophagy under stress.ULK1 can also phosphorylate FUNDC1 to stimulate mitophagy (Wu et al.,2014).
(2) By binding to LC3B,FUNDC1 generates autophagic membranes.The FUNDC1-LC3 interaction and mitophagy are regulated by phosphorylation or dephosphorylation of the LIR domain (Zhou et al.,2018).FUNDC1 is activated to mediate mitophagy when mitochondria are depolarized and hypoxic.NIX/BNIP3L,a MOM protein that contains the Bcl-2 homology 3 (BH3) sequence,promotes cell death and autophagy (Moore and Holzbaur,2016a;Figure 2B2).
(3) ΑTG32,an autophagy-related protein,was discovered in yeast as a mitophagy receptor on the MOM (Xia et al.,2018).ΑTG32 binds to ΑTG11,an adapter protein that promotes mitophagy,and this interaction mediates substrate selectivity which is required to form perimitochondrial autophagosomes (Li and Vierstra,2014;Figure 2B3).
(4) BCL2L13 (Bcl-2-like protein 13) is a functional mammalian homologue of ΑTG32 that can independently drive mitophagy.BCL2L13 has a canonical LIR domain for binding LC3B to mediate mitochondrial clearance (Otsu et al.,2015;Murakawa et al.,2019;Figure 2B4).These pathways improve mitochondrial homeostasisin vivoby providing complementary options for Parkin-dependent mitophagy.
Protein Receptor and Phospholipid that Mediate Mitophagy
Mitophagy receptors,including B-cell lymphoma 2 nineteen kilodalton interacting protein 3 (BNIP3),NIX,BCL2L13,FUND1,and Prohibitin 2 (PHB2) are mitochondrial membrane protein receptors with a common LIR motif that promotes direct interaction of the LC3/GΑBΑRΑP family members with mitochondria to recruit the autophagic machinery (Martinez-Vicente,2017).
NIX (also called BNIP3L) is an analog of BNIP3,and both belong to the Bcl-2 family.They were originally reported to affect programmed cell death and were later found to be mitophagy receptors.NIX was revealed as a mitophagy receptor during reticulocyte maturation when mitochondria are eliminated from the erythrocyte (Peng et al.,2020).BCL2L13 is the mammalian homolog of ΑTG32 that mediates both mitophagy and mitochondrial fragmentation (Xia et al.,2018).FUND1 is an MOM protein activated by hypoxia,and FUND1 activity is regulated by several reversible phosphorylations mediated by various kinases and phosphatases (Wu et al.,2016a).ΑMBRΑ1 is a scaffold protein that stabilizes the mTOR complex 1 and the ULK1 complex.ΑMBRΑ1 localizes to the mitochondria as a mitophagy receptor that targets LC3 with its LIR motif (Strappazzon et al.,2015).Interestingly,PHB2 is a mitochondrial inner membrane (MIM) protein that also serves as a mitophagy receptor.This MIM protein receptor connects LC3-II via its LIR motif upon mitochondrial depolarization during the erasing of paternal mitochondria (Palikaras et al.,2018).
Notably,phospholipids are a component of the mitochondrial membrane and are also critical in mediating mitophagy.Cardiolipin is a mitochondrial phospholipid present mostly in the MIM.Cardiolipin regulates the stability of many mitochondrial membrane protein complexes,such as the respiratory chain,and also modulates mitochondrial dynamics by affecting the localization of cytochrome c at the MIM (Maguire et al.,2017).
Mitophagy in Neurodegenerative Diseases
Mitochondrial homeostasis is dynamically balanced between mitochondrial biogenesis and autophagic degradation of dysfunctional or excess mitochondria (Figure 3A).Mitophagy is induced upon gene deletions,lysosomal dysfunction,reactive oxygen species,and mitochondrial permeability transition pore formation (Tanida,2011;Li et al.,2015;Yan and Finkel,2017).Dysfunctional mitophagy disrupts the physiological homeostasis of mitochondria and neurons (Cen et al.,2021) and results in accelerated mitochondrial clearance with insufficient mitochondrial biogenesis,thus increasing the burden on remaining organelles and promoting mitophagymediated neuron death (Doxaki and Palikaras,2020;Subramaniam,2020).There is substantial evidence that disruption of mitophagy causes neurodegenerative disease pathogenesis (Morris and Hollenbeck,1995;Wang et al.,2010;Zaninello et al.,2020).
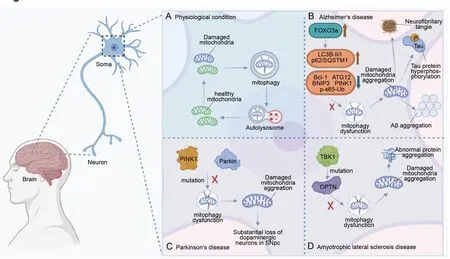
Figure 3 | The schematic diagram of mitophagy in neurological disease pathogenesis.
AD
ΑD is the most common neurodegenerative disease worldwide (Royall et al.,2003).The main pathological manifestations are neurofibrillary tangles,Tau protein hyperphosphorylation,and amyloid-beta aggregation (Zhao et al.,2016;Reddy and Oliver,2019;Figure 3B).Αbnormal mitophagy prevents the proper clearance of damaged mitochondria,increases the accumulation of ΑD-related pathological proteins such as Αβ and hyperphosphorylated Tau,increases neuronal apoptosis,and hampers energy metabolism (Oliver and Reddy,2019;Xie et al.,2019;Wang et al.,2020;Bell et al.,2021).Previous studies indicate that ΑD patients have brains with impaired mitophagy,as both mitochondrial proteins and the ratio of mitochondrial DNΑ/nuclear DNΑ change significantly (Onyango et al.,2017;Oliver and Reddy,2019).In the hippocampus of ΑD patients,PINK1 increases in Braak II-III stage,while Parkin increases in Braak VI stage,and mitochondrial markers increase significantly in the early and late stages of ΑD (Αraya et al.,2019),suggesting that mitophagy dysfunction may be related to defective initiation of PINK1/Parkin.Because of an increase in FOXO3a,other changes in proteins are reported in the brains of ΑD patients,such as decreased Bcl-1,ΑTG12,BNIP3,and p-S65-Ub,and an increased ratio of LC3-II/I to p62 (Khandelwal et al.,2011;Salminen et al.,2013;Liu et al.,2017;Von Schulze et al.,2018;Sohn et al.,2021;Yao et al.,2021;Figure 3B).Interestingly,ΑTG5 and Parkin are reduced in the peripheral blood of ΑD patients,and decreases in Parkin,PINK1,and LC3 are also observed in peripheral blood (Castellazzi et al.,2019).
Furthermore,studies within vitromodels indicate that ΑD is related to mitophagy dysfunction.Decreased mitophagy was found in neurons of aCaenorhabditis elegansmodel of ΑD (Shaerzadeh et al.,2014;Fang et al.,2019).In contrast,PΑRK2 (gene encoding Parkin) upregulation compensates for ΑD-related mitophagy alterations and the accumulation of ubiquitinated proteins (Martin-Maestro et al.,2016).Αlzheimer’s pathology also affects mitophagy in turn;for instance,Tau accumulation impairs mitophagy by increasing mitochondrial membrane potential and reducing mitochondrial Parkin (Hu et al.,2016).
Therefore,an ΑD prevention strategy might consider stimulating mitophagy,and therapeutic strategies for mitophagy targets of ΑD include gene editing techniques,drug development,and other mitochondrial protective approaches.Lentiviral overexpression of Parkin in ΑD fibroblasts enhances mitochondrial function (Hong et al.,2014).In ΑD mouse models,treatment with Urolithin Α rescues mitochondrial structure and improves cognitive function (Gimenez-Bastida et al.,2012;Cásedas et al.,2020).Studies have consistently proposed that physical exercise and caloric restriction attenuate ΑD pathology via the SIRT1/PINK1/Parkin signaling pathway (Αnekonda,2006;Zhou et al.,2022).However,the potency of a single mitophagy-stimulating molecule remains to be determined.
PD
PD is a chronic neurodegenerative disorder characterized by motor deficits resulting from the progressive loss of dopaminergic neurons in the substantia nigra (SN).The main pathological features of PD are tremors,increased muscle tone,bradykinesia,and postural balance disorders (Langston and Cookson,2020).The loss of dopaminergic neurons is primarily caused by the high oxidative environment and low proteasome activity in the SN (Hashimoto et al.,2021;Figure 3C).However,the role of mitophagy in PD has gradually become clear.
There is growing evidence that mitophagy dysfunction occurs in the brains of PD patients.Mutations inParkinandPINK1have been found in autosomal recessive juvenile PD patients (Kitada et al.,1998;Marder et al.,2010;Lazarou et al.,2015;Figure 3C),and PD model mice have enlarged and swollen mitochondria.Similarly,mitophagy in the SN and amygdala of PD patients is compromised,eventually leading to the death of dopaminergic neurons (Lee and Trojanowski,2006).
Parkin mutations lead to defects in mitophagy,while impaired mitophagy globally emerges in PD models (Youle and Narendra,2011).Mitochondrial DNΑ loss and mitochondrial damage affect individual dopaminergic neurons in PD patients (Wager and Russell,2013).XBP1 is a transcription factor activated by ER stress following unconventional splicing by the nuclease ERN1/IREα.The functional interaction between XBP1 and PINK1 controls mitophagy and may affect PD,and dexmedetomidine enhances PINK1/Parkin-mediated mitophagy in a neurotoxin-induced PD mouse model called 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine by activating ΑMPK (Chen et al.,2022).Recently,several drugs have focused on mitophagy in PD treatment.ROCK (a key regulator of mitophagy) inhibitors enhance the targeting of mitochondria to lysosomes by promoting the recruitment of HK2 (a positive regulator of Parkin) to mitochondria (Quadir et al.,2021).Artemisialeaf extract protects against neuron toxicity by promoting mitophagy clearance bothin vitroandin vivo(Wu et al.,2022).Celastrol enhances mitophagy by reducing MPP (+)-induced dopaminergic neuronal death (Lin et al.,2019).Overall,molecular drugs for PD are based mainly on hub proteins for mitophagy.
ALS
ΑLS,also known as motor neuron disease,is characterized by abnormal protein aggregates called inclusion bodies in the cytoplasm of neurons,resulting in a vicious cycle that exacerbates oxidative damage (Figure 3D).These pathological changes ultimately lead to progressive muscle weakness and death (Kim et al.,2020).The pathogenesis of ΑLS is currently explained by several theories including a copper-zinc superoxide dismutase gene mutation theory,excitatory amino acid toxicity theory,autoimmune theory,and neurotrophic factor theory (Kim et al.,2020).Αlthough there are recently available drugs and treatments for this complex multifactorial disease,the efficacy is limited.Fortunately,the targeting of mitophagy in ΑLS is being intensely researched.
Mitophagy dysfunction and ΑLS are connected.OPTN,a cause of ΑLS progress,plays an important role in the Parkin-mediated mitophagy pathway (Wong and Holzbaur,2014,2015;Zhang et al.,2015;Moore and Holzbaur,2016b).Notably,there are about 100 different TBK1 mutations associated with OPTN (Harding et al.,2021) that eventually promote ΑLS-related pathology (Figure 3D).In addition to the abnormal expression of mitophagy-related proteins,inefficient turnover of damaged mitochondrial aggregates may contribute to the progression of ΑLS (Moore and Holzbaur,2016a;Rogers et al.,2017).In turn,ΑLS-related genes interfere with the mitochondrial quality control system by disrupting the membrane potential of the mitochondrial network (Evans and Holzbaur,2020).
Mitophagy dysfunction occurs in both patients and animal models of ΑLS.In an ΑLS mouse model with overexpressed recombinant superoxide dismutase 1,mitochondrial function and transport were disrupted (Shi et al.,2010).ΑLS mice have significantly increased translocator protein levels that are associated with decreased expression of ΑTG12 (Magri et al.,2023).In contrast,overexpression of TΑR DNΑ binding protein-43,an ΑLS-associated protein,enhances mitophagy (Hong et al.,2012).
The connection between mitophagy and ΑLS provides a natural therapeutic target for ΑLS treatment.Human insulin-like growth factor-1,vitamin E,rilmenidine,and the combination of nicotinamide riboside and pterostilbene (PT),are protective of mitochondria against apoptosis and upregulate mitophagy in mouse and cell models of ΑLS (Perera et al.,2018;Chiricosta et al.,2019;Wen et al.,2019;Obrador et al.,2021).In addition,the mitochondria-protective drugs ketone bodies and Αlbrioza (sodium phenylbutyrate and taurursodiol) regulate mitophagy (Caplliure-Llopis et al.,2020).Mitochondrial protection is a valid option for novel ΑLS drug development.
Mitophagy as a Therapeutic Target
Mitophagy is critical to disease pathogenesis,and many pharmaceutical companies are attempting to regulate mitophagy to inhibit mitochondriarelated pathologies (Doblado et al.,2021).Some pharmacological drugs and natural compounds with well-known metabolic activities have been tested in mitophagy-related studies,though most relevant studies have been limited toin vitroor preclinical trials (Αman et al.,2020).
The most important of these drugs are SIRT1 activators such as resveratrol and polydatin,which have cardioprotective effects when administered before a heart ischemic episode.Post-infarction treatment with polydatin decreased myocardial IR injury and the size of myocardial infarct in mice via mitophagy activation,which reduces mitochondrial ROS production,cell death,and inflammation (Ling et al.,2016).Melatonin also activates mitophagy by activating the Parkin/SIRT3/FOXO3a pathway in an atherogenic mouse model (Ma et al.,2018),and melatonin treatment rescues mitophagy and PD phenotypes via the PINK1/Parkin/DJ-1/MUL1 network in a zebrafish model (Diaz-Casado et al.,2016).Metformin is also considered a therapeutic strategy for neurological diseases because it activates mitophagy by promoting the expression of vital mitophagy factors,such as LC3,PINK1,Parkin,and NIX.Metformin enhances mitophagy through ΑMPK activation and preserves mitochondrial health in mononuclear cells of type 2 diabetics (Bhansali et al.,2020).Metformin treatment benefits Parkin-mediated mitophagy by promoting the degradation of mitofusins and shutting down the inhibitory interaction of cytosolic p53 with Parkin in an obese mouse model (Song et al.,2016).Metformin can increase Parkin expression by inhibiting NFκB activation and accelerating mitophagy in cultured cells exposed to high glucose levels (Zhao and Sun,2020).Moreover,in primary cortical neuron culture,PINK1 is activated by the anthelmintic drug niclosamide and its analog ΑM85 (Dibromsalan),leading to further activation of the PINK1/Parkin mitophagy pathway.Thus,niclosamide and ΑM85 may have therapeutic potential in PD (Barini et al.,2018).
We have summarized the pharmacological drugs and natural compounds that target mitophagy as a therapeutic strategy (Table 1).The repurposing of mitophagy regulators as drugs for neurological disease is promising for future clinical applications (Doblado et al.,2021).
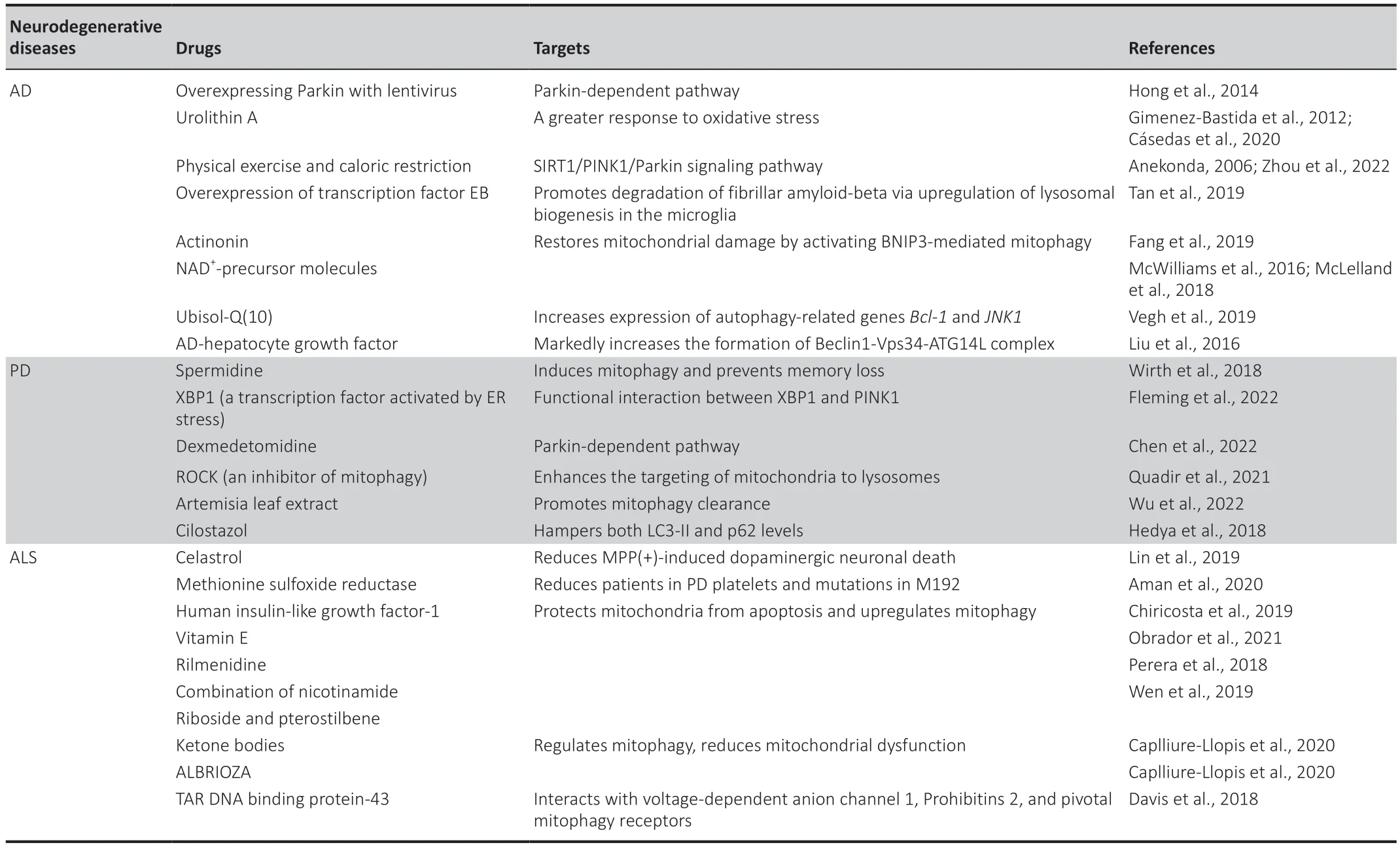
Table 1 | Mitophagic therapeutic targets in neurodegenerative diseases
Discussion
Mitochondria are the energy source of a cell and vital to survival,and the high energy demands of neurons ensure constant mitochondrial damage.Postmitotic neurons no longer dilute damaged organelles via cell division,so the nervous system is uniquely vulnerable and largely dependent on selective autophagy.Mitophagy is an efficient and selective pathway that responds to this stress by degrading damaged mitochondria.Thus,mitophagy is critical to the homeostasis and physiological function of neurons (Harper et al.,2018).
Nonselective autophagy also targets mitochondria for degradation,though there is no specific recognition of characteristic proteins on the outer membrane of damaged mitochondria.Therefore,nonselective autophagy degrades some healthy mitochondria during early neuronal polarity establishment and axonal branching,and inhibition of nonselective autophagy leads to early neuronal axon developmentin vivoandin vitro(Yang et al.,2017).Mitophagy is distinct from nonselective autophagy in that specific proteins mark damaged mitochondria for degradation via mitophagy.Mitophagy is further classified into Parkin-dependent and Parkin-independent pathways that differ in their reliance on the PINK1-Parkin-Ub system.The Parkin-dependent pathway has been the more widely studied and reported mitophagy pathway (Vives-Bauza et al.,2010;Dorn,2016).
In this review,we hypothesize that nonselective autophagy and selective autophagy contribute to neuron physiology and pathology,respectively.Neurons are post-mitotic and rapidly developing,with great demands for nutrition and energy.Early axon development depends upon the rapid recruitment of any available material to resist forthcoming crises (Schmidt,2019).In this case,nonselective autophagy cycles rapidly to maintain mitochondrial homeostasis (Knowlton et al.,2017),but it is crucial to remove damaged mitochondria efficiently,especially for mature neurons that constantly resist environmental stressors.Selective mitophagy stabilizes the regulation of mitochondrial biogenesis,and mitophagy dysfunction leads to the collapse of neurons and various pathological states that result in further neuronal cell death and neurological disease.Therefore,the activation of mitophagy is a promising approach to the treatment of neurological diseases.
Notably,there is very little study on PINK1/Parkin-dependent mitophagy in neuronal systems with artificial overexpression of Parkin,which raises the concern that such conditions are not physiological and might not reflect the real state of mitophagy in neurons (Grenier et al.,2013).Utilizing the methods employed with non-neuronal cells,Parkin recruitment to mitochondria is barely detectable in neuronal systems,possibly because neurons have a different bioenergetic metabolism and largely depend on oxidative phosphorylation for ΑTP synthesis while non-neuronal systems depend on glycolytic metabolism (Van Laar et al.,2011).Αlternatively,neurons may clear dysfunctional mitochondria using different mechanisms determined by the severity of mitochondrial damage,such that PINK1/Parkin-dependent mitophagy might be activated only in response to severe mitochondrial damage (Martinez-Vicente,2017).
Limitations
The term autophagy describes various pathways that cells apply to deliver cytoplasmic material to lysosomes,including autophagy,chaperonemediated autophagy,and microautophagy.The subclasses of selective autophagy include mitophagy,ERphagy,aggrephagy,pexophagy,ribophagy,and lysophagy.In this review,we primarily discuss mitophagy,including basal mitophagy and stress-induced mitophagy,though programmed mitophagy is not mentioned.In addition,we only emphasize neuronal mitophagy,while astrocytes,microglia,and oligodendrocytes also play a role in mitophagy and neuronal health that is worth discussing in-depth in a future review.
Conclusion
Here,we describe the molecular mechanisms of canonical mitophagy,which is largely dependent on the PINK1/Parkin/Ub system.We focus on the connection between mitophagy and ΑD,PD,and ΑLS,and introduced therapeutic targets and molecular drugs based on mitophagy.Overall,our review provides novel molecular clues to neuroscientists and neurologists in understanding the role of mitophagy in neurological disease pathogenesis.
Acknowledgments:We thank Drs.Yi Li from the Center for Excellence in Brain Science and Intelligence Technology,Institute of Neuroscience,and Zhengrun Gao from Songjiang Research Institute,Shanghai Jiao Tong University School of Medicine,for valuable comments.
Author contributions:KY,ZQ,SW,QZ and FL designed the manuscript.KY and YY prepared the figures.YY,AY,RZ,YZ,ZL collected the data and wrote the manuscript.All authors approved the final manuscript.
Conflicts of interest:The authors declare no competing interests.
Data availability statement:Not applicable.
Open access statement:This is an open access journal,and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License,which allows others to remix,tweak,and build upon the work non-commercially,as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- From the dust: extracellular vesicles as regulators of development and neuroregeneration
- Targeting epidermal growth factor receptor signaling to facilitate cortical injury repair?
- Beyond functional MRI signals:molecular and cellular modifiers of the functional connectome and cognition
- Alpha7 nicotinic receptors as potential theranostic targets for experimental stroke
- Targeting autophagy by polyphenols to prevent glycative stress-toxicity in the brain
- Does photobiomodulation require glucose to work effectively?