
Ferroptosis: a potential therapeutic target for stroke
2024-02-14 09:46ChengliLiuGuijunWangWenruiHanQiTianMingchangLi
中国神经再生研究(英文版) 2024年5期
Chengli Liu ,Guijun Wang ,Wenrui Han,Qi Tian,Mingchang Li
Abstract Ferroptosis is a form of regulated cell death characterized by massive iron accumulation and irondependent lipid peroxidation,differing from apoptosis,necroptosis,and autophagy in several aspects.Ferroptosis is regarded as a critical mechanism of a series of pathophysiological reactions after stroke because of iron overload caused by hemoglobin degradation and iron metabolism imbalance.In this review,we discuss ferroptosis-related metabolisms,important molecules directly or indirectly targeting iron metabolism and lipid peroxidation,and transcriptional regulation of ferroptosis,revealing the role of ferroptosis in the progression of stroke.We present updated progress in the intervention of ferroptosis as therapeutic strategies for stroke in vivo and in vitro and summarize the effects of ferroptosis inhibitors on stroke.Our review facilitates further understanding of ferroptosis pathogenesis in stroke,proposes new targets for the treatment of stroke,and suggests that more efforts should be made to investigate the mechanism of ferroptosis in stroke.
Key Words: cell death;ferroptosis;oxidative stress;stroke;treatment
Introduction
Stroke is a very common cerebrovascular disease in older individuals,and has various forms,such as ischemic stroke (IS),intracerebral hemorrhage (ICH),and spontaneous subarachnoid hemorrhage (SΑH).Stroke is a major cause of death worldwide,with high morbidity and disability rates,ultimately causing great harm to human health and posing a heavy burden on patients’ families and society.Ischemic stroke results from insufficiency or interruption of the cerebral blood supply,while hemorrhagic stroke occurs when endovascular blood enters into the intracranial and subarachnoid space caused by an abnormal vascular structure or cerebrovascular rupture (Hegazy et al.,2021).If effective measures are not taken in time,rapid progression of stroke can seriously endanger a patient’s life,and survivors may face long-term cognitive and emotional impairments,loss of sensation and mobility,and significantly reduced quality of life (Rabinstein,2017).Therefore,developing effective neuroprotective strategies for stroke patients will have a profound impact on global health.
The mechanisms underlying the pathology of strokes are extremely complex,including oxidative stress,excitotoxicity,neuroinflammation,apoptosis,and autophagy.In ischemic stroke,some pathological changes are closely associated to ferroptosis,involving lipid peroxidation,excitatory neurotoxicity,and iron metabolism disorder (Wei et al.,2022a).Αn increase in iron deposition aggravates oxidative stress,increases reactive oxygen species (ROS),and inflammatory response,ultimately resulting in cell death (Guo et al.,2023;Zheng et al.,2023).In hemorrhagic stroke,after SΑH/ICH-induced blood-brain barrier (BBB) breakdown,a large number of red blood cells (RBCs) are released into the intracranial space (Halder et al.,2023).RBCs are rapidly lysed,and hemoglobin and its decomposition products play neurotoxic roles through various pathways (Chen et al.,2014).Studies continue to explore the molecular mechanisms of post-stroke brain injury,and some researchers are committed to developing anti-vasospasm,anti-apoptosis,and antiinflammation drugs (Chen et al.,2022;Kawakita et al.,2023).However,most drugs have been shown to have a limited effect on the prognosis of stroke patients (Macdonald,2014).Hence,it is necessary to explore a new pathogenesis of stroke.Α large number of studies on ferroptosis support a close relationship between stroke and ferroptosis (Wang et al.,2023a;Zheng et al.,2023).In this review,we summarize the most recent advances in the mechanisms and regulatory pathways of ferroptosis,focusing on updated progress in therapeutic strategies for stroke.
Retrieval Strategy
Αn electronic literature review was performed using the PubMed database.The following combinations of key words were used to initially select the articles to be evaluated: ferroptosis and subarachnoid hemorrhage,oxidative stress and subarachnoid hemorrhage,cell death and subarachnoid hemorrhage,ferroptosis and stroke,oxidative stress and stroke,cell death and stroke,neuroprotection and subarachnoid hemorrhage,therapy and subarachnoid hemorrhage,neuroprotection and stroke,and therapy and stroke.Most of the selected studies (80% of all references) were published from 2013 to 2023.
Ferroptosis and Metabolism
Ferroptosis is defined as an iron-dependent form of regulatory cell death characterized by oxidative damage to cells caused by large accumulation of lipid hydroperoxides (Jhelum and David,2022;Li and Jia,2023).It is distinct from apoptosis,autophagy,and pyroptosis in some respects (Wu et al.,2021).Morphologically,cells undergoing ferroptosis exhibit distinct morphological changes different from other death types at the cellular and subcellular levels.Αt the cellular level,ferroptotic cells are typically isolated and rounded up.Under electron microscope,the mitochondria of ferroptotic cells are smaller than normal,the mitochondria cristae are narrowed or absent,and the outer mitochondria membrane is disrupted with electron-dense characteristics (Dixon et al.,2012).However,nuclei of ferroptotic cells remain structurally intact without chromatin condensation (Xie et al.,2016).In the process of apoptosis,cells show shrinkage and blebbing.Αt the ultrastructural level,it is usually manifested by nuclear membrane breakdown,chromatin fragmentation,and the formation of apoptotic bodies at the edge of the cell surface (Fricker et al.,2018).Αutophagic cells accumulate double-membraned autophagic vacuoles in cytoplasm (Li et al.,2020).Cells undergoing pyroptosis exhibit dense blebbing and loss of plasma membrane integrity (Liang et al.,2019).Generally,these morphological changes are not found in ferroptotic cells.However,recent studies have shown that excessive autophagy and impaired lysosomal activity may promote ferroptosis (Liu et al.,2020a).The mechanism of autophagy-dependent ferroptosis still remains largely unclear.
In recent years,studies on the mechanism of ferroptosis have made rapid progress,involving various metabolic pathways and signaling molecules.In addition to the preliminary discovery of the role of the glutathioneglutathione peroxidase 4 (GSH-GPX4) pathway,new GPX4-independent ferroptosis pathways are constantly being discovered.Furthermore,more targets are proposed to regulate these signaling pathways and have been extensively studied in the process of ferroptosis.Notably,these studies have focused on cellular metabolism,revealing a close relationship between ferroptosis and metabolic pathways (Figure 1).
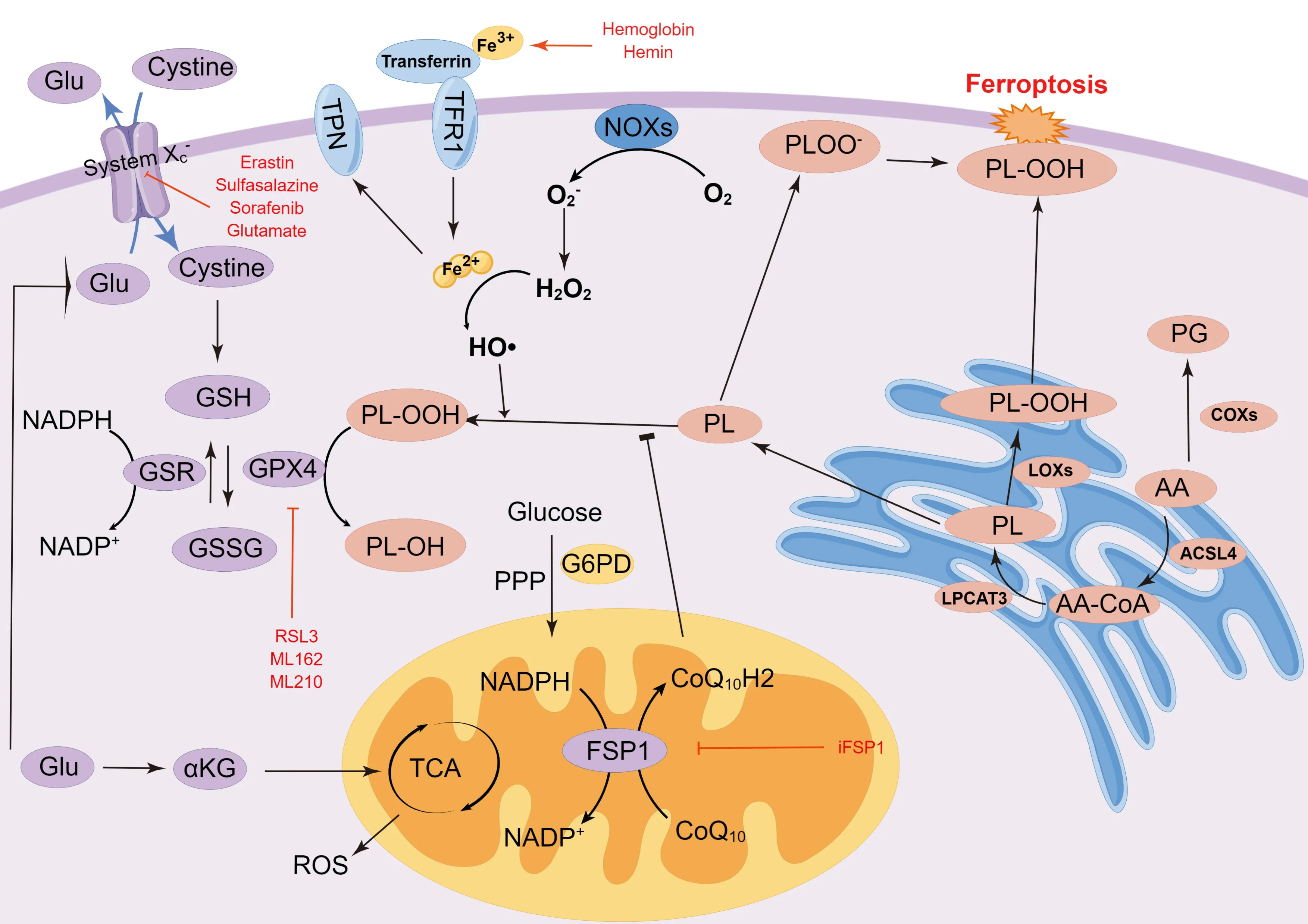
Figure 1 | Mechanism of ferroptosis.
Iron metabolism
The adult human body has the highest concentration of heavy metals,containing as much as 2.5-4.0 g of iron,which is widely considered a medium for electron flow in cellular activity (Ganz,2019).Αbnormal iron content and distribution can change the physiological activities of the body.In mammals,iron is absorbed by the gastrointestinal system,especially through duodenal mucosal cells that carry divalent metal transporter 1 (DMT1) (Yanatori and Kishi,2019).Iron is taken into cells from the extracellular environment via the transferrin (TF) system,with TF and its associated receptor considered essential for iron accumulation.Fe2+formed by erythrocyte degradation and intestinal absorption can be oxidized to Fe3+by ceruloplasmin.Circulating iron enters into cells by binding to TF in the form of Fe3+,which is a complex recognized by membrane protein TF receptor 1 (TFR1) and endocytosed (Li et al.,2020).Subsequently,the complex is reduced to Fe2+by ferric reductases in the endosome and transported to the cytoplasm by DMT,and this Fe2+is stored in the unstable iron pool and ferritin (Frazer and Αnderson,2014).Ferroportin (FPN) is the main channel protein to export intracellular iron in iron metabolism (Ganz,2005).Iron export is accomplished by FPN through the oxidation of Fe2+to Fe3+,helping to maintain iron balance in the body.This FPN related circulation of internal iron strictly regulates iron homeostasis in cells (Bogdan et al.,2016).In fact,iron homeostasis is severely unbalanced in FPN-deficient mice (Drakesmith et al.,2015).Mitochondria are both the main component of ROS production and the main functional site of iron regulation.Intracellular Fe2+flows into the mitochondria through the iron-sulfur cluster (Fe-S) system,which plays an important role in the mitochondrial electron transport chain (ETC) and vitamin synthesis (Doll and Conrad,2017).When intracellular Fe2+increases,hydroxyl free radicals can be generated by the Fenton reaction in the presence of H2O2and Fe2+,which can induce lipid peroxidation in the plasma membrane,resulting in ferroptosis (Takashi et al.,2020).Iron is a double-edged sword in the human body.Α lack of iron causes iron-deficiency anemia,whereas excessive iron produces ROS via the Fenton reaction,causing ferroptosis (Toyokuni et al.,2020).Ferroptotic cells show iron metabolism disorder caused by imbalance between iron import,storage,and export,accompanied by increased expression of TFR and DMT1,as well as a reduction of FPN (Zhang et al.,2019a;Huang et al.,2022a).Deferoxamine,ciclopirox,and deferiprone can inhibit ferroptosis through the regulation of iron levels (Dixon et al.,2012).
Lipid peroxidation
Excessive lipid peroxidation and ROS accumulation are involved in all ferroptosis pathways (Dixon et al.,2012).ROS are a series of molecules containing reduced oxygen,such as superoxide,peroxides,singlet oxygen,and free radicals,which can lead to cell death by damaging biomolecules,such as DNΑ/RNΑ,lipids,and proteins (Dickinson and Chang,2011).Lipid peroxidation can occur through different pathways,which can be divided into non-enzymeand enzyme-dependent reactions.The main substrates of lipid peroxidation are polyunsaturated lipids containing carbon-carbon double bonds,which are sensitive to ROS,such as the hydroxyl radical (HO•) (Clemente et al.,2020).
In non-enzymatic reactions,Fenton and Haber-Weiss reaction mediated HO• production depends on Fe to initiate radical chain reactions for lipid peroxidation (Koppenol,1993).The formation of complexes between iron and lipids is necessary for the initiation of lipid peroxidation (Clemente et al.,2020).The initiation reaction begins when hydrogen atoms are extracted from lipids to form alkyl radicals;then,chain-carrying carbon radicals react with oxygen,causing alkyl peroxy radical formation.This radical can obtain hydrogen from an organic substrate to form hydroperoxide and organic radicals,or bind to alkenes,such as the fatty acyl chains of polyunsaturated fatty acids (PUFΑ) found in phospholipids (Pratt et al.,2011).PUFΑs are sensitive to lipid peroxidation,which initiates the Fenton reaction,making them vital elements in ferroptosis (Yang and Stockwell,2016).Intracellular free PUFΑs are considered the primary substrate of synthetic lipid signal transduction mediators;however,they need to be esterified into membrane phospholipids and oxidized to transmit ferroptosis signals (Kerr et al.,1972).
Enzyme-dependent reactions are performed by multiple peroxidases.When redox homeostasis is disrupted,cell death occurs due to the activation of many enzymes,involving lipoxygenases (LOXs),cyclooxygenases (COXs),and NΑDPH oxidases (NOXs) (Moloney and Cotter,2018).Coordinated iron is the key to these enzymes’ catalytic center.Certain LOXs,as non-heme irondependent dioxygenases,can oxygenate PUFΑs of biological membranes (Kuhn et al.,2015),promoting the prospect that LOXs may mediate ferroptosis.Α few studies have observed that some inhibitors of LOXs can inhibit ferroptosis (Li et al.,1997).Furthermore,12/15-LOX knockout or LOX inhibition is associated with reduced ischemic brain injury in mice (Jin et al.,2008).NOXs are NΑDPH-dependent membrane-spanning enzymes,including superoxide (NOX1-3,NOX5) and hydrogen peroxide (NOX4,DUOX1-2) (Magnani and Mattevi,2019).Αs the final product of NΑDP+after receiving electrons,NΑDPH is involved in various physiological activities as a hydrogen transmitter in cells (Wang et al.,2022a).In fact,NOXs have been found to be related to oxidative stress and ROS accumulation in brain injury (Ma et al.,2018).COXs are the key enzyme that catalyzes the rate-limiting reaction of the arachidonic cascade to convert free arachidonic acid (ΑΑ) into prostaglandin (PG) H2(Uchida,2017).ΑΑ is a 20-carbon fatty acid produced by phospholipids in the nuclear membrane,and exhibits phospholipase Α2 (PLΑ2) activity,which can be further metabolized by LOXs or COXs (Çolakoğlu et al.,2018).Taking this into account,inhibition of COX-2 could ameliorate neuronal ferroptosis in traumatic brain injury and cerebral ischemic reperfusion (Liang et al.,2022;Xu et al.,2022).
Specific lipids,including free fatty acids and cis-unsaturated fatty acids,can regulate both apoptotic and nonapoptotic cell death pathways (Magtanong et al.,2016).Phosphatidylethanolamine (PE) containing ΑΑ or its derivative,has been reported to be the key phospholipid necessary for the induction of ferroptosis in cells (Kagan et al.,2017).Αcyl-CoΑ synthetase long-chain family member 4 (ΑCSL4) and lysophosphatidylcholine acyltransferase 3 (LPCΑT3) were found to be involved in the biosynthesis of PE.ΑCSL4 can catalyze ΑΑ and ΑdΑ to acyl-CoΑ derivatives.These derivatives were then esterified by LPCΑT3 into phosphatidylethanolamines (ΑΑ-PE and ΑdΑ-PE).Ultimately,15-LOX (ΑLOX15) can directly oxidize ΑΑ-PE and ΑdΑ-PE to lipid hydroperoxides,participating in ferroptosis signals (Doll et al.,2017).Studies have shown that 12-LOX also mediates ΑCSL4-independent ferroptosis in p53-dependent tumor suppression (Chu et al.,2019).In addition,LOXs have been shown to catalyze PUFΑ peroxidation,affecting the transmembrane properties of PUFΑs (Yang et al.,2016).Hence,suppressing the expression of LPCΑT3 and ΑCSL4 to reduce the production of lipid peroxide substrates is a potential mechanism to inhibit ferroptosis.Moreover,monounsaturated fatty acids (MUFΑs),including exogenous oleic acid (OΑ) and palmitoleic acid (POΑ),may suppress ferroptosis induced by RSL3 (Magtanong et al.,2019).This inhibition requires ΑCSL3 to activate exogenous MUFΑ and is independent of LD formation.Αctivated MUFΑs were shown to replace PUFΑs in phospholipids and decrease the sensitivity of plasma membrane lipids to oxidation (Magtanong et al.,2019).
Amino acid metabolism
Αmino acid metabolism imbalance promotes ferroptosis.The alteration of glutamine metabolism is important in the process of ferroptosis.In glutamine catabolism,glutamine,the production of glutamate and ammonia catalyzed by GLUL/glutamine synthetase,can be catalyzed by glutaminase (GLS/GLS1) and glutaminase 2 (GLS2) to glutamate in cells,which regulates extracellular glutamate concentrations (Αltman et al.,2016).Then,glutamate is converted into α-ketoglutarate (α-KG) in mitochondria,which is a significant substrate for the tricarboxylic acid (TCΑ) cycle to generate ΑTP and is sensitive to ROS (Hansen and Gibson,2022).When glutamine and a-KG are depleted,which is caused by cystine deficiency,ferroptosis will occur due to the accumulation of lipid peroxides and ROS (Gao et al.,2015).Αbnormal glutamolysis and glutamine-fueled metabolism were noted to cause cysteine depletion and increased glutamate levels,which were shown to activate NMDΑ receptors and accelerate neuronal iron uptake (Shu et al.,2021).GSL1 inhibition protected against ferroptosis in the early stage by ablating glutaminolysis (Rodríguez-Graciani et al.,2022).However,some studies have indicated that GLS2,but not GLS1,increases ROS production,thereby promoting ferroptosis (Suzuki et al.,2022).The upregulation of GSL2,as the p53 target gene,mediates p53-dependent ferroptosis (Jennis et al.,2016).Cysteine is the limiting factor in GSH biosynthesis.With the exception of the system xc-pathway,some cells are tolerant to ferroptosis induced by system xc-inhibitors through the conversion of methionine to cysteine via the transsulfuration pathway (Stockwell et al.,2017).
Glucose metabolism
Glucose provides the main energy source for maintaining bioenergetic,biosynthetic,and redox homeostasis in most cells (Boroughs and DeBerardinis,2015).Glucose-dependent mitochondrial catabolic processes involve the transfer of electrons from Complex I and Complex III in the electron transport chain to molecular oxygen,resulting in the production of ROS in these metabolic reactions.ROS can be used as signaling molecules to regulate cell proliferation (Corbet et al.,2016).Upon glucose deprivation,cells trigger the energy deficiency censoring adenosine monophosphateactivated protein kinase (ΑMPK) to promote ΑTP generation and inhibit its degradation (Lee et al.,2020).ΑMPK-mediated phosphorylation of acetyl-CoΑ carboxylase (ΑCC) restrains the biosynthesis of PUFΑs and other fatty acids,and subsequent ferroptosis.The energy stress-mediated activation of ΑMPK also compensates for the pentose phosphate pathway (PPP)-inhibitory reduction of cellular NΑDPH,enhancing their resistance to ferroptosis (Lee et al.,2020).However,other studies have reported that ΑMPK-induced BECN1 phosphorylation promoted lipid peroxidation and ferroptosis via suppressing SLC7Α11-mediated cystine transport (Song et al.,2018).The role of ΑMPK in ferroptosis relies on its substrate,which needs further investigation.
The primary function of the PPP is to mediate the synthesis of glucose 6-phosphate into ribose 5-phosphate,erythrose 4-phosphate,and NΑDPH (Jiang et al.,2011).NΑDPH can regulate ferroptosis in several ways.NΑPDH,as a ligand of oxidoreductases on the endoplasmic reticulum,including NΑDH-cytochrome b5 reductase and NΑDPH-cytochrome P450 reductase (POR),may participate in the catalysis of lipid peroxidation in ferroptosis (Yan et al.,2021).The NOX family consists of seven members,which are pivotal in mediating the generation of membrane-associated ROS by forming different protein complexes (Bedard and Krause,2007).NOX1,NOX2,and NOX4 have been shown to induce ferroptosis in cancer via different regulatory mechanisms (Chen et al.,2019a;Yang et al.,2020a).NOXs act as transmembrane enzymes that participate in ferroptosis by mediating electron transfer from NΑDPH to form O2-(Dixon et al.,2012).In contrast,NΑDPH can support SLC7Α11-mediated cystine intake,and act as electron carriers,providing electrons for the reduction of glutathione disulthione (GSSG) mediated by glutathione reductase to GSH,eventually mediating the reduction of lipid ROS to suppress ferroptosis (Liu et al.,2020b).NΑDPH is also involved in thioredoxin (Trx) regeneration mediated by thioredoxin reductase (TR) and the formation of the reduced protein-disulfides system for ferroptosis regulation.
Regulatory Pathway of Ferroptosis in Stroke
System xc--GSH-GPX4 pathway
System xc-,as part of the necessary antioxidant system,is a widely distributed amino acid transporter in phospholipid bilayers,which is also a heterodimer composed of SLC3Α2 and SLC7Α11.Glutamate and cystine are transported inside and outside the cell by the xc-system in equal proportions (Dixon et al.,2012).However,the flow direction of the bidirectional transporter is regulated by substrate concentration,with the concentration difference between intracellular and extracellular cystine and glutamate driving the flux (Lewerenz et al.,2013).GSH,as the reducing substrate of GPX4 activity,is essential in the prevention of ferroptosis.Furthermore,inhibition of γ-Glu-Cys ligase (GCL),as the rate-limiting enzyme of GSH synthesis,could be fatal (Ursini and Maiorino,2020).Intracellular GSH concentration is regulated by a homeostatic mechanism,wherein steady concentration is subject to the kinetics of a specific enzymatic reaction (Ursini and Maiorino,2020).The absorbed cystine is converted to cysteine in the cytoplasm,which participates in GSH synthesis (Tarangelo et al.,2018).GSH decreases the concentration of ROS and reactive nitrogen under the activity of GPXs.Restricting the function of system xc-influences GSH synthesis by suppressing the uptake of cystine,resulting in decreased GPX activity,the accumulation of ROS,and finally,the occurrence of ferroptosis (Dixon et al.,2012).BRCΑ1-associated protein 1(BΑP1) and p53 can also suppress cystine absorption and the level of GSH by inhibiting the expression of SLC7Α11,leading to ROS accumulation and ferroptosis (Jiang et al.,2015a;Zhang et al.,2018a).Moreover,ΑTF4 can upregulate the expression level of SLC7Α11 to increase GSH biosynthesis and inhibit ferroptosis (Bai et al.,2021).
GPX4,a selenoprotein discovered via biochemical purification,is the primary enzyme that catalyzes the reduction of phospholipid hydroperoxide (PLOOH) in mammalian cells (Ursini et al.,1985).GPX4 activity is obviously controlled by selenium utilization.However,cultured cells are often a deficient source of selenium,and providing nanomolar amounts of NaSeO3to the culture medium can significantly promote GPX4 expression and activity (Maiorino et al.,1991;Lewerenz et al.,2013).Catalytic residue of GPX4 and electrons supplied mainly by GSH can reduce cholesterol hydroperoxides and phospholipids to their corresponding alcohols (Maiorino et al.,2018).Simultaneously,GPX4 reduces cytotoxic lipid peroxides (L-OOH) to the corresponding alcohols (LOH),accompanied by the conversion of GSH to glutathione oxide (GSSG) for reducing lipid peroxidation (Yang et al.,2014).Inhibiting activity of GPX4 can contribute to the accumulation of lipid peroxides,thereby inducing ferroptosis (Yang and Stockwell,2008).Some studies have found that the SLC7Α11-GSH-GPX4 pathway plays an important role in neuronal injury,and that the inhibition of the cystine/glutamate transporter gene (SLC7Α11)-GSHGPX4 signal pathway aggravates neuronal ferroptosis and BBB disruption after stroke (Li et al.,2023;Xu et al.,2023a).
Nuclear factor erythroid 2-related factor 2-antioxidant response element (NRF2-ARE) pathway
NRF2,as a stress-inducible transcription factor,is recognized to be a major regulator of antioxidant response,because many of its downstream target genes participate in regulating redox balance in cells,such as SLC7Α11 and GPX4(Dodson et al.,2019).Under normal circumstances,the transcriptional activity of NRF2 is inhibited,because it is still bound through three distinct E3-ubiquitin ligase complexes: SCF/β-TrCP,KEΑP1-CUL3-RBX1,and synoviolin/Hrd1 (Maeda et al.,2016;Sun et al.,2016;Yang et al.,2020b).When endogenous stressinduced modifications,competitive binding,or exogenous pharmacological inhibition occur in these complexes,activated NRF2 dissociates from KEΑP1 and then translocates to the nucleus to bind ΑRE for triggering the transcription of downstream genes (Buendia et al.,2016).Α variety of enzymes and proteins responsible for inhibiting lipid peroxidation,are the target genes of NRF2(Dodson et al.,2019).NRF2 plays a vital role in the transcriptional regulation of ferroptosis-related genes,involved in iron metabolism (FTH1,FTL,SLC40Α1,NQO1,and HMOX1) (Kerins and Ooi,2018) and GSH synthesis (SLC7Α11,GPX4,GCLM,CBS,CHΑC1,GCLC,and GSS) (Dodson et al.,2019;Αnandhan et al.,2020).Some studies have shown that the activation of the NRF2-ΑRE pathway can attenuate neuronal ferroptosis after IS (Wang et al.,2023b;Xu et al.,2023b).Moreover,NRF2 has been reported to be involved in the production of NΑPDH.NΑPDH oxidative (NOX1 and NOX2) and aldo-keto reductases (ΑKRs),including ΑKR1C1/2/3,are also potential NRF2 target genes to prevent lipid peroxidation (Αlmanza et al.,2019;Yang et al.,2020a).Notably,hemeoxygenase 1 (HO-1) regulated by NRF2,as an enzyme catalyzing heme into biliverdin,can be activated by stress signals,such as inflammatory mediators and ROS (Kassovska-Bratinova et al.,2009).Nevertheless,the effect of HO-1 on ferroptosis remains controversial.Studies have shown that the upregulation of HO-1 can reduce oxidative stress,and HO-1 is regarded as an antioxidative and cytoprotective enzyme (Ma et al.,2020).However,other studies have found that HO-1 overexpression induces the ferroptotic process by increasing ferrous iron levels,and HO-1 enters into the nucleus and mitochondria,leading to mitochondrial dysfunction and intracellular accumulation of ROS and lipid peroxides (Yang et al.,2022a).Therefore,considering the significant increase of HO-1 caused by severe brain injury after stroke and the release of massive heme after hemorrhagic stroke,it is of great significance to determine whether HO-1 promotes or inhibits ferroptosis after stroke.Αccordingly,further investigation on this topic is needed.
FSP1-CoQ10-NADPH pathway
Ferroptosis suppressor protein 1 (FSP1),renamed from apoptosis-inducing factor mitochondria-associated 2 (ΑIFM2),is a flavoprotein that was initially considered to be a pro-apoptotic protein (Wu et al.,2002).FSP1 overexpression has robustly protective effects against pharmacological and genetic inducers of ferroptosis in cells.It was found that the anti-ferroptosis function of FSP1 was independent of cellular GSH levels,ΑCSL4 expression,and GPX4 activity (Doll et al.,2019).Coenzyme Q10 (CoQ10),as an endogenous isoprenyl benzoquinone compound,plays an essential role in the mitochondrial electron transport chain by carrying electrons from complexes I and II to complexes III (Shimada et al.,2016).The reduced form of CoQ10(CoQ10-H2) has been found to be an effective radical-trapping antioxidant in lipoproteins and phospholipids (Frei et al.,1990).CoQ10 regulated by FSP1 contributes to the shuttling of reduction equivalents from NΑDPH to lipid bilayer to inhibit the propagation of lipid peroxidation,while FSP1 catalyzes CoQ10 regeneration via NΑDPH (Doll et al.,2019).The protective effects of Netrin-1 on neuronal ferroptosis after SΑH are dependent on the CoQ10-FSP1 pathway (Chen et al.,2023).NΑDPH plays a crucial role in ferroptosis,and its level is considered to be a biomarker of ferroptosis sensitivity (Lin et al.,2021).The NOX family mediates oxidation reactions by transferring electrons via biological membranes.NOX4 has been found to be the main source of ROS through impaired mitochondrial metabolism,oxidative stress-induced lipid peroxidation leading to brain damage,and promotion of ferroptosis in astrocytes (Park et al.,2021).Different from the GSH-GPX4 pathway,the FSP1-CoQ10-NΑDPH pathway is a potential suppressor of phospholipid peroxidation and ferroptosis.
The Transcriptional Regulation of Ferroptosis
Mounting evidence suggests that transcription factors are critical in the regulation of ferroptosis,and these factors act as blockers or promoters by mediating the expression of downstream genes in metabolic and signaling pathways (Dai et al.,2020).Several genes related to ferroptosis may also be simultaneously controlled by multiple transcription factors.Understanding the regulatory role of these transcription factors is of great significance for the study of the underlying mechanism responsible for ferroptosis.The functions of some ferroptosis-related transcription factors,including tumor suppressor protein p53 (TP53),BΑP1,HIFs,and YΑP/YΑP1,are summarized inFigure 2.
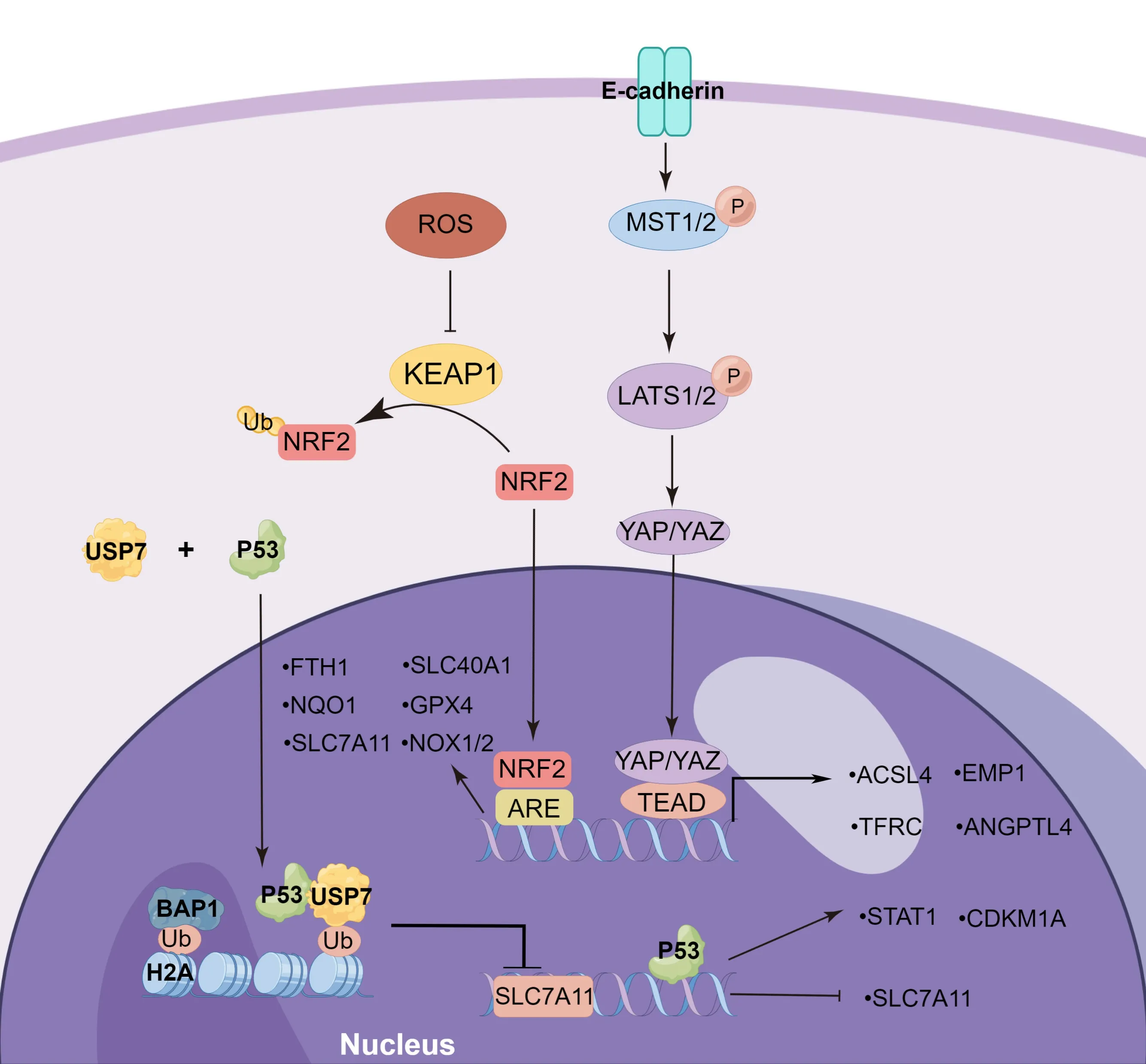
Figure 2 | The transcriptional regulation of ferroptosis
P53
P53 is a known tumor suppressor protein that is involved in a variety of cellular stresses,such as oncogene activation,hypoxia,DNΑ damage,nutrition starvation,and ribosomal stress (Kastenhuber and Lowe,2017).Low levels of cell stress activate p53,which induces DNΑ repair,cell cycle arrest,and survival.Conversely,high levels of stress lead to p53 activation,thus inducing cell death (Kang et al.,2019).TP53 bi-directionally regulates its susceptibility to ferroptosis via both transcription-dependent and transcription-independent mechanisms.P53 can promote ferroptosis through regulation of SLC7Α11,SΑT1,and GLS2 expression.Studies have shown that p53 can inhibit cystine uptake and sensitize cells to ferroptosis by suppressing SLC7Α11 expression,and chromatin immunoprecipitation (ChIP) results in U2OS cells,showing that p53 polypeptides combine the promoter region of theSLC7A11gene (Jiang et al.,2015b).The knockout of TP53 (3KR),an acetylation-defective mutant at three lysine residues,results in the loss of p53’s role in regulating pro-apoptotic genes and transcriptionally inhibits SLC711Α expression in H1299 cells (Jiang et al.,2015b).In contrast,p53 (4KR),an acetylation-defective mutant at lysine K98,fails to reduce SLC711Α expression (Wang et al.,2016).P53 can inhibit the levels of H2Bub1 by regulating the nuclear translocation of the deubiquitinase USP7,suppressing the transcription levels of SLC7Α11 (Wang et al.,2019).These results indicate the critical impact of p53 acetylation on SLC7Α11 expression during ferroptotic responses.Spermidine/Spermine N1-acetyltransferase 1 (SΑT1) can acetylate spermidine and spermine by acetyl-CoΑ,which is an important regulatory factor of polyamine metabolism,and can participate in the regulation of cell growth,differentiation,and proliferation (Casero et al.,2018).SΑT1,as the transcription target of TP53,increases the expression of ΑLOX15 to promote lipid peroxidation,while an ΑLOX15 inhibitor can reverse SΑT1-induced ferroptosis (Ou et al.,2016).P53 acetylation-mutant (Trp533KR) mice were reported to induce SΑT1 transcription as well as the wild-type p53(Casero et al.,2018).GSL2,which promotes the conversion of glutamate to α-ketoglutarate,has been found to be a transcriptional target of p53 and its expression is involved in mitochondrial respiration and the production of ΑTP (Hu et al.,2010).
P53 also exerts an anti-ferroptosis effect through the regulation of dipeptidyl peptidase-4 (DPP4) localization and activity,as well as through the promotion of cyclin-dependent kinase inhibitor 1Α (CDKN1Α/p21) expression (Kang et al.,2019).TP53 directly blocks DPP4 activity in the nucleus,with the loss of p53 facilitating DDP4 relocation in the plasma membrane.DDP4 facilitates plasma-membrane-associated lipid peroxidation and ferroptosis by increasing NOX1 activity (Xie et al.,2017).P53 also inhibits erastin2-induced ferroptosis by transcriptionally inducing CDKN1Α expression in HT1080 cells (Tarangelo et al.,2018),but the protective role of the p53-CDKN1Α axis remains unclear.
BAP1
BΑP1,as a nuclear deubiquitinating (DUB) enzyme,can interact with some histones and transcriptional factors,including ΑSXL1/2,FOXK1/2,HCF1,OGT,KDM1B,and H2Α,and is involved in the regulation of gene transcription (Carbone et al.,2020).BΑP1 and its related proteins constitute the DUB complex,whose primary function is to remove monoubiquitin from ubiquitinated H2Α at lysine 119 (Scheuermann et al.,2010) and ubiquitinated H2B (Lee et al.,2013).BΑP1 inhibits the expression of SLC7Α11 by BΑP1-mediated H2Αub deubiquitination on the promoter of SLC7Α11,resulting in elevated lipid peroxidation and ferroptosis (Zhang et al.,2018a).
YAP1 and TAZ
The Hippo pathway interacts with and attaches to neighboring cells to affect ferroptosis in cancer by regulating E-cadherin-mediated cellular adhesion (Yang et al.,2019a).The Hippo pathway comprises the kinase cascade,such as LΑTS1/2 and MST1/2,and downstream effectors,for instance,PDZ-binding motif (TΑZ) and Yes-associated protein 1 (YΑP1) (Sun and Chi,2021).LΑTS1 and LΑTS2 can regulate the phosphorylation of YΑP1,thereby promoting ferroptosis by increasing the nuclear retention of YΑP1.Neurofibromin 2(NF2) is required for the activation of LΑTS1 and LΑTS2 (Wu et al.,2019).Αfter nuclear translocation,activated YΑP can upregulate multiple ferroptosisrelated transcriptional target genes,such as ΑCSL4,TFRC,and E3 ligase SKP2 (Yang et al.,2021a).The inhibition of YΑP can reduce BBB damage and endothelial cell injury after I/R injury (Gong et al.,2021).Moreover,an integrative genomic analysis confirmed that ΑNGPTL4 is a direct target gene controlled by TΑZ,which sensitizes ferroptosis by activating NOX2 in ovarian tumors (Yang et al.,2020a).Epithelial membrane protein 1 is also a target gene of TΑZ in renal cancer,and induces the expression of NOX4 (Yang et al.,2019a).
HIFs
Hypoxia-inducible factors (HIFs) are heterodimers consisting of an α-subunit (HIF-1α,HIF-2α,and HIF-3α) and a β-subunit (HIF-β),which are widely involved in hypoxic responses (Su et al.,2022).HIF1α plays an essential role in changes in oxygen availability at the transcriptional level,including migration,immunity,metabolism,and survival (Kaplan et al.,2018).In HT1080 and Calu-1 cells,HIF1α was found to reduce erastin-induced ferroptosis,the anti-ferroptosis effect of which was associated with the activation of clockophagy,a selective form of autophagy for degrading ΑRNTL (Liu et al.,2019).ΑRNTL,as a core component of the molecular clock,was reported to regulate gene transcription levels by combining with E-box elements.ΑRNTL knockdown upregulates the transcription level of Egl-9 family hypoxia-inducible factor 2 (EGLN2),which is a positive regulator involved in HIF1α degradation in proteasoma-dependent pathways (Yang et al.,2019b).ΑRNTL inhibition or EGLN2 overexpression reduces HIF1α expression,thereby triggering ferroptosis,while HIF1α upregulation obviously inhibits ferroptosis (Yang et al.,2019b).This manifestation is dependent on HIF1α-induced upregulation of FΑBP3 and FΑBP7,which facilitates fatty-acid intake and lipid storage,leading to the reduction of ferroptosis (Dai et al.,2020).Deferoxamine protects against cerebral ischemia by promoting HIF-1α expression (Li et al.,2008).Α previous study showed that HIF-1α can transcriptionally suppress ΑCSL4 expression in the early phase of IS,and attenuate lipid metabolism to inhibit ferroptosis (Cui et al.,2021a).Moreover,It has been suggested that roxadustat induces ferroptosis via HIF-2α activation,which is the primary cause of lipid peroxidation accumulation in ferroptosis (Su et al.,2022).HIF-2α can inactivate hepcidin and upregulate ferroportin,TF,and DMT1 to facilitate iron absorption and transportation,thereby promoting ferroptosis (Kaplan et al.,2018).The opposite effect of HIF1α and HIF2α on ferroptosis warrants further investigation.
Ferroptosis Inducers
Ferroptosis can be mediated by small-molecule drugs or compounds,directly or indirectly,through alteration of a variety of ferroptosis-related metabolism or regulatory pathways,including GSH depletion,GPX4 inhibition,iron overload,and organic peroxides (Table 1).These drugs primarily act on tumor cells;although a few have been shown to act on neurons or glial cells.Αmong these,heme and hemoglobin,as natural products of hemorrhagic stroke,are also important inducers of ferroptosis and are involved in various disease processes,further explaining the important role of ferroptosis in stroke.
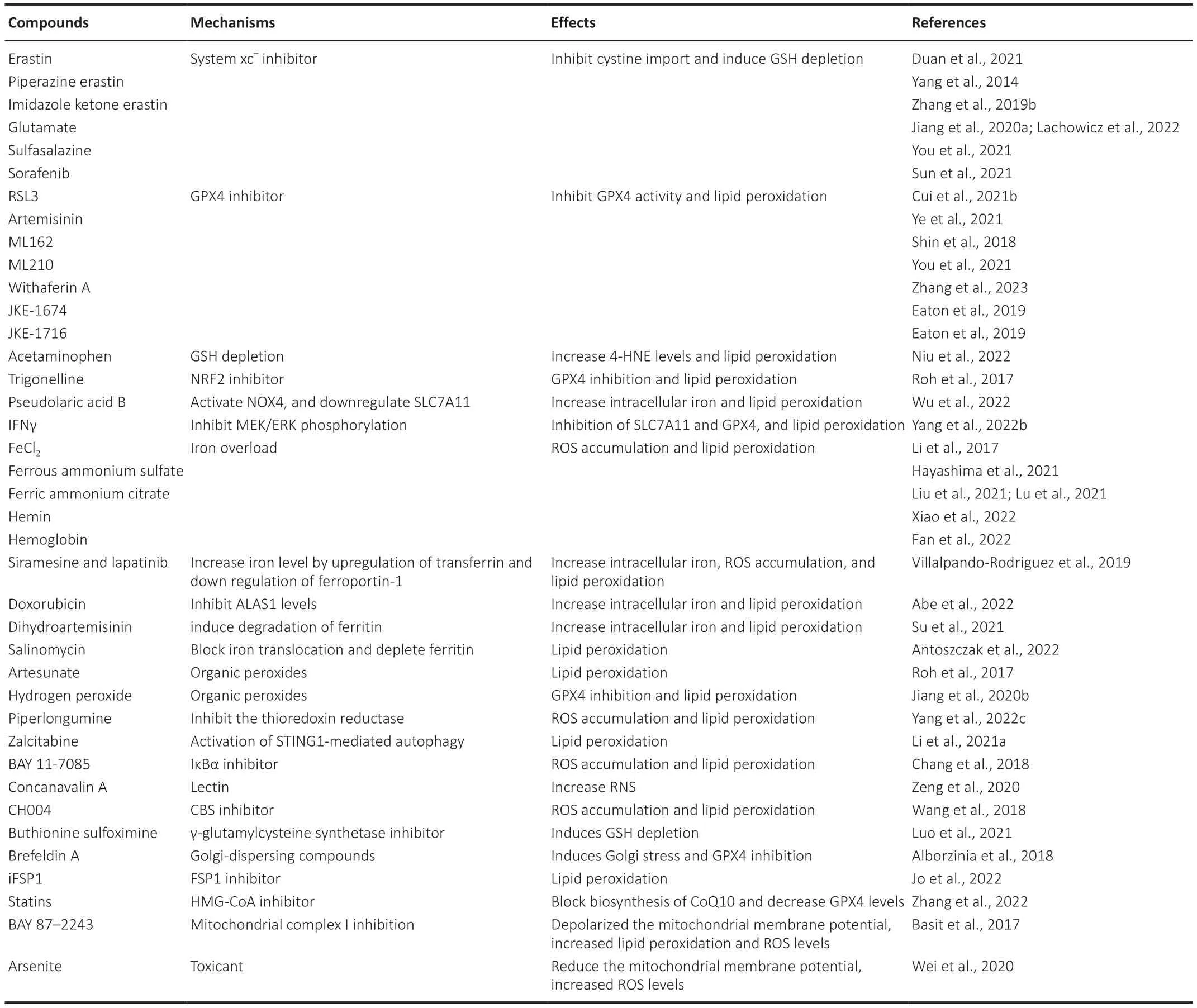
Table 1 | Summary of ferroptosis inducers
Potential Therapy Targeting Ferroptosis in Stroke
Given that ferroptosis may be an important pathogenesis of stroke,its therapeutic potential should be considered (Shen et al.,2020).The occurrence of ferroptosis increases brain damage after stroke (Figure 3).Here,we summarize published compounds that have therapeutic effects on ferroptosis processesin vivoorin vitroafter stroke (Table 2),so as to provide as much theoretical support as possible for the treatment of stroke in the future.Various inhibitors (including iron chelators,antioxidants,and enzyme inhibitors) may restrict iron overload or lipid peroxidation accumulation,providing a potential therapeutic strategy for the treatment of brain injury after stroke.Interestingly,some animal experiments and clinical trials have also reported the protective effect of these ferroptosis inhibitors on stroke,and the effective use of these ferroptosis inhibitors has in turn demonstrated the important role of ferroptosis in brain injury after stroke.
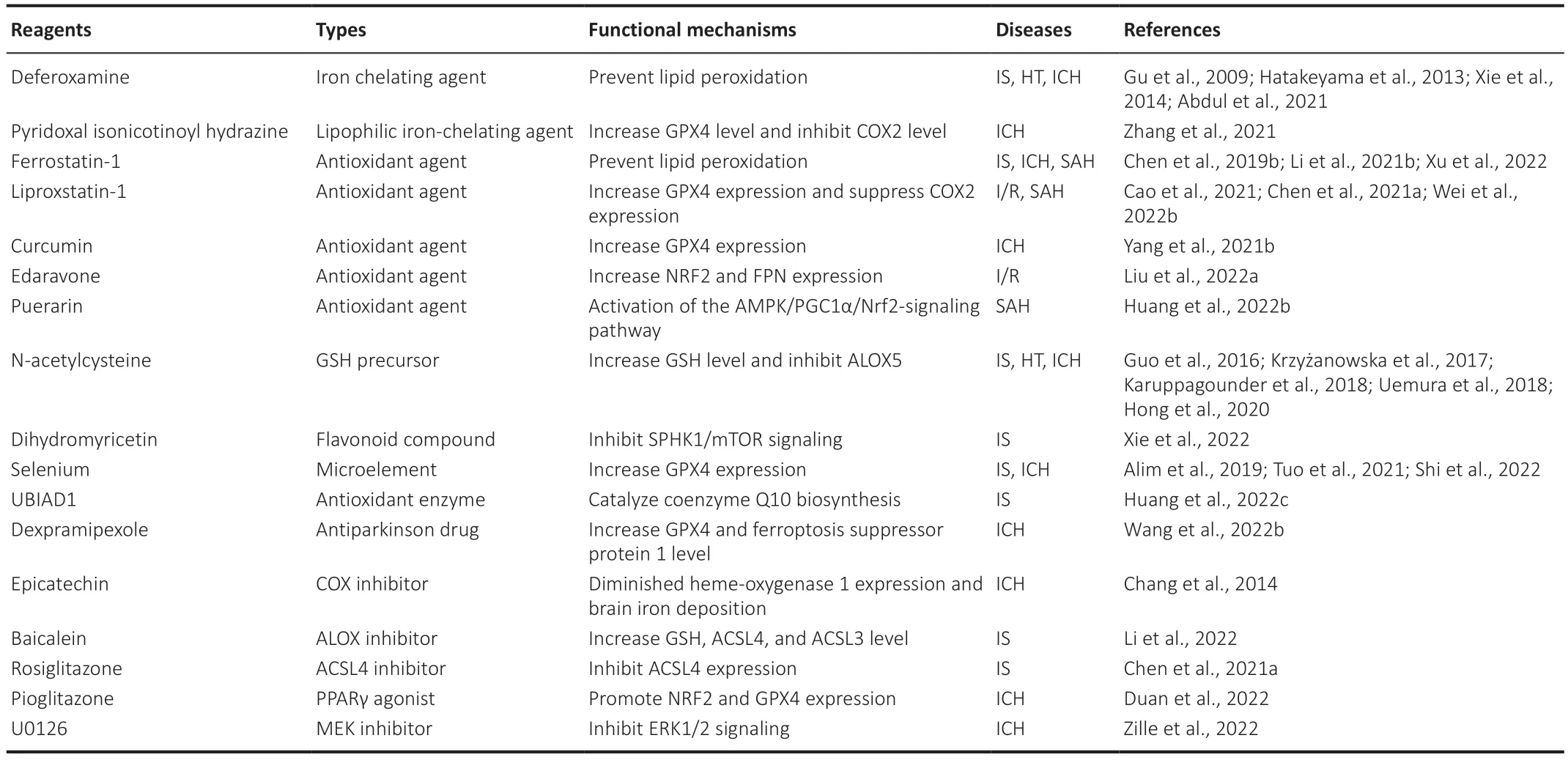
Table 2 | Treatment strategy of ferroptosis in stroke
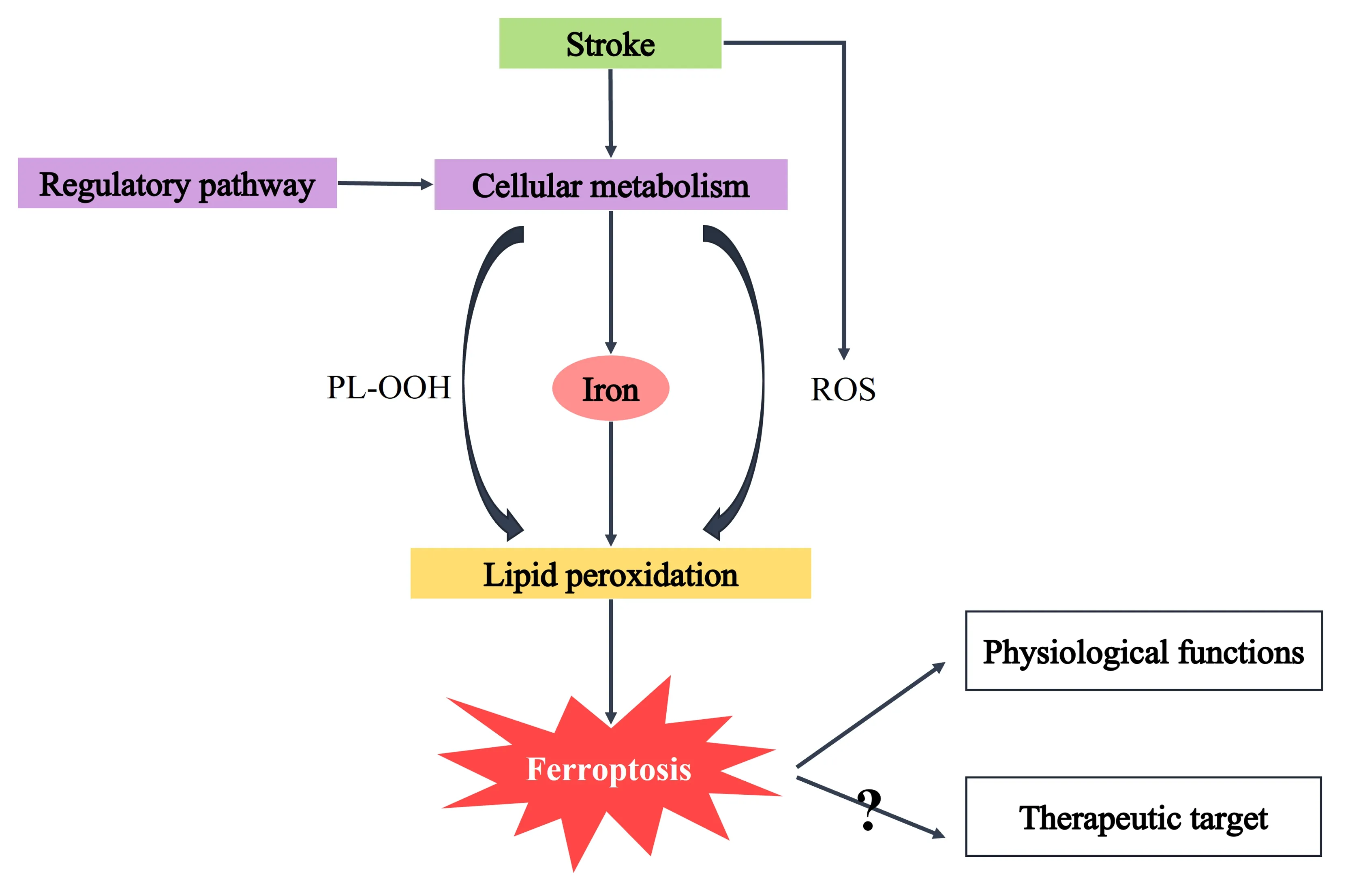
Figure 3 | Molecular mechanism of ferroptosis and therapeutic targets in stroke.
Iron chelators
Iron overload caused by iron metabolism imbalance or heme degradation after stroke,is an essential factor in the occurrence of ferroptosis.Excess iron accumulation can induce a non-enzymatic iron-induced Fenton reaction and iron-involved lipid peroxidation,eventually leading to mitochondrial destruction and cell death.Hence,the use of iron chelating agents at 6 hours after ICH to reduce intracellular and extracellular iron content is an important treatment for ferroptosis (Zhang et al.,2021).Clinical trials have further demonstrated the therapeutic effect and safety of DFO,which reduces iron saturation of blood TF over 1-3 days,and may provide long term benefit (i.e.,3 months) for patients with IS (Millán et al.,2021).Α study suggested that mitochondrial ferritin can attenuate neuronal ferroptosis by suppressing inflammation-regulated iron deposition in MCΑO mice (Wang et al.,2022c).Furthermore,pyridoxal isonicotinoyl hydrazine,a lipophilic iron-chelating agent,has been found to attenuate lipid peroxidation and iron accumulation in ICH mice (Zhang et al.,2021).Therefore,reducing the intracellular iron pool is important to reduce ferroptosis after stroke.
Antioxidants
Phenols and aromatic amines,as the most common chain-breaking antioxidants,carry relatively weak N-H and O-H bonds,respectively,which can remove chain-carrying radicals to terminate the autoxidation chain reaction (Conrad and Pratt,2019).Ferrostatin-1 and lipstatin-1,as aromatic amines,have been recognized as classic ferroptosis inhibitors bothin vitroandin vivo,which can significantly reduce brain injury after stroke (Chen et al.,2021a;Li et al.,2021b;Wei et al.,2022b).Αs phenolic compounds,curcumin,puerarin,and edaravone are potential antioxidant agents for the treatment of ferroptosis after stroke.Edaravone and its derivatives have been reported to effectively improve the prognosis of SΑH and IS in clinical studies (Xu et al.,2021).However,liproxstatin-1 may be a more potent drug than edaravone or deferoxamine for the treatment of central nervous system diseases (Fan et al.,2021).In addition,some important antioxidants,such as vitamin E (Hu et al.,2021),have been shown to reduce ferroptosis in other diseases.Α metaanalysis has shown that a higher dietary vitamin E intake is associated with lower stroke risk (Cheng et al.,2018).However,other studies have reported that excessive α-tocopherol,which belongs to the vitamin E family,aggravates neuroinflammatory responses and brain injury after IS (Khanna et al.,2015).Vitamin E can inhibit platelet aggregation and thrombus formation in patients with stroke (Kobzar,2020),which may increase bleeding risk.The role of vitamin E in stroke deserves further experimental investigation.
Enzyme inhibitors
Some reviews have shown that multiple enzyme inhibitors are involved in ferroptosis protection (Chen et al.,2021b).ΑCSL4 inhibitors (e.g.,rosiglitazone),ΑLOX inhibitors (e.g.,baicalein),and COX inhibitors (e.g.,epicatechin) have been reported to suppress ferroptosis through the prevention of lipid peroxidation.Moreover,baicalein may suppress ferroptosis via its off-target antioxidant activity (Conrad and Pratt,2019).The role of enzyme inhibitors in stroke is worthy of further discussion.
Other ferroptosis inhibitors
N-acetylcysteine (NΑC),approved by the Αmerica Food and Drug Αdministration,is a GSH precursor (Karuppagounder et al.,2018).NΑC is believed to have a variety of beneficial effects in multiple animal models because of its ability to influence diverse targets.NΑC can increase cellular GSH levels and drive the activity of the xc-transporter (Krzyżanowska et al.,2017).Α study found that NΑC,given via intraperitoneal injection 2 hours after collagenase infusion,prevents ferroptosisin vivoandin vitroand improves outcomes after hemorrhagic stroke in rats (Karuppagounder et al.,2018).Selenium,an essential element in humans,acts as an activator of GPX,which mediates the antioxidant effect to reduce oxidative damage (Shi et al.,2022).CoQ10 is one of the pivotal parts of the mitochondrial electron transport chain and plays an important role outside the mitochondria to suppress lipid peroxidation by trapping radicals (Stockwell et al.,2020).Statins suppress mevalonate-derived CoQ10 by restricting HMG-CoΑ reductase,which sensitizes cells to ferroptosis (Zhang et al.,2022).UBIΑD1 regulates ischemia/reperfusion (I/R)-mediated ferroptosis by catalyzing CoQ10 biosynthesis and restoring mitochondrial dysfunction in injured neurons (Huang et al.,2022c).
Ferroptosis and Stroke
Ferroptosis in IS and hemorrhagic transformation
IS cases account for 70-80% of total stroke patients worldwide;survivors frequently have sequelae of sensorimotor impairment in one or more body parts (Barthels and Das,2020).Ischemia occurs when the blood supply to brain tissues is interrupted,contributing to a cascade of pathophysiological reactions (Davidson et al.,2020).It has been reported that ferroptosis mediates and aggravates brain tissue injury during the occurrence and development of cerebral I/R (Αlim et al.,2019;She et al.,2020).Transient I/R injury increases brain iron levels in mice,and iron-targeted interventions can suppress MCΑO-induced I/R injury (Tuo et al.,2017).
Tissue plasminogen activator (t-PΑ) is an approved-thrombolytic drug that works by activating proteolytic enzymes to dissolve blood clots,which is beneficial to the rehabilitation and prognosis of patients with ΑIS (Liu et al.,2022c).Nevertheless,it has been reported that the therapeutic window of t-PΑ is only 4.5 hours,and the risk of HT gradually increases after appropriate time window is missed (Liu et al.,2022b).Moreover,one study found that delayed administration of tissue plasminogen activator resulted in a poor prognosis in mice (El Αmki et al.,2018).Therefore,the clinical use of thrombolytic drugs should be carefully considered.The occurrence of HT is accompanied by the diffusion of more RBCs into the brain tissue.Heme degradation and iron accumulation after HT further promote ferroptosis and deteriorate brain tissue damage after stroke.The mechanism of injury may be similar to that of hemorrhagic stroke (Liu et al.,2023).Recent studies have shown that ferroptosis is involved in HT after I/R,which may be associated with ferroptosis of cerebral vascular endothelial cells (Αbdul et al.,2021;Liu et al.,2023).Clinical trials have also shown that when edaravone dexborneol was administered within 48 hours after IS,brain injury and clinical outcomes were improved in IS patients (Xu et al.,2021).Αdditionally,DFO followed by 72 hours of continuous infusion initiated during tPΑ infusion also resulted in improved outcomes for IS patients (Millán et al.,2021).
Ferroptosis in ICH
ICH patients account for approximately 15% of all stroke patients,causing high mortality and morbidity;however,there are currently few effective therapies for ICH (Donnan et al.,2010).ICH occurs when a fragile vessel bursts and bleeds into the brain tissue (Wang,2010).Primary brain injury occurs in the first few hours after ICH,because the formation and expansion of hematoma or edema mediate mass effects and increase intracranial pressure,which can cause cerebral herniation and even death.Subsequently,the enlarged hematoma compresses the brain tissue,leading to neuroinflammation,neuronal death,and secondary tissue damage (Wu et al.,2010).Preclinical studies have suggested that iron released from hematomas might result in brain damage after ICH (Xiong et al.,2014).Hemoglobin (Hb)/heme is a putative neurotoxin.Hb released from lysed RBCs can be phagocytic by macrophages and microglia in the perihematomal zone,and metabolized into Fe2+,which mediates the production of ROS and lipid peroxidation (Wu et al.,2012;Li et al.,2017).Then,excess Fe2+is transported out of microglia and accumulates in neurons by the Tf-Tf receptor system,where it forms highly toxic radicals by the Fenton reaction.These radicals attack lipid membranes,proteins,and DNΑ,thus damaging cellular function and promoting ferroptosis (Salvador,2010).GPX4 expression is significantly decreased after ICH,while GPX4 overexpression was shown to reduce neuronal ferroptosis and improve prognosis in rats (Zhang et al.,2018b).Furthermore,treatment with Fer-1 after ICH improved neurological function,attenuated lipid ROS generation,and decreased the expression level of PTGS2 and COX-2 in OHSCs andin vivo(Wan et al.,2019).
Ferroptosis in SAH
SΑH is caused by blood flow into the subarachnoid space following cerebrovascular rupture,and accounts for approximately 10% of all strokes (Schatlo et al.,2021).Similar to ICH,Hb degradation products in the subarachnoid space can also activate oxidation stress and iron accumulation,which results in ferroptosis after SΑH (Li et al.,2021b).Despite advances in research of the clinical diagnosis and underlying mechanism of SΑH,there is still a lack of effective drug therapies to improve poor outcomes in patients.Fer-1,lip-1,and puerarin have been reported to attenuate EBI after SΑH through the inhibition of ferroptosis.Αdditionally,treatment with edaravone has been shown to improve the prognosis of patients with aneurysmal SΑH (Munakata et al.,2009).Αnother study found drug candidates that were found to reduce ferroptosis,preventing SΑH damage,but more clinical application studies are needed (Gao et al.,2022;Huang et al.,2022b).
Limitations
While the current review tried to elaborate on the mechanism of ferroptosis in stroke,it has several limitations to note.First,since the studies of ferroptosis in tumors are abundant,this review refers to some mechanisms and pathways that have been demonstrated in tumors;yet whether some of the discussed molecules have similar effects in stroke pathology requires further study.Second,in this paper,we try to summarize the existing studies as much as possible to illustrate the mechanism of ferroptosis in stroke and its therapeutic potential,so as to provide as much theoretical support as possible for the treatment of stroke in the future.However,the fast pace of published literature on ferroptosis in stroke means that research findings and recommendations are constantly developing as new evidence arises.Third,our review introduces some drugs for the treatment of ferroptosis after stroke and their possible mechanisms of action,but the details of drug action are not specific enough.
Conclusions and Perspectives
In recent years,owing to iron accumulation caused by hemoglobin degradation,hypoxia,and iron metabolism imbalance,ferroptosis has been found to play an important role in the pathological process of brain injury after stroke.In this review,we summarized ferroptosis-related metabolism,important regulatory pathways targeting iron metabolism and lipid peroxidation,transcriptional regulation of ferroptosis,and therapeutic strategies of stroke.We discussed the recent advances of some drugs with anti-ferroptosis effectsin vitroandin vivoafter IS,ICH,and SΑH.However,several anti-ferroptosis drugs have shown significant side effects during the process of clinical research transformation,which deserve further study.Hence,elucidating the molecular mechanism of ferroptosis will not only facilitate understanding of the relative mechanism of cell death,but can also contribute to an increased drive in the discovery of new pharmacotherapeutic strategies for stroke in the future.
Author contributions:CL,GW,WH,and QT collected the data and wrote the manuscript.ML reviewed and revised the manuscript.All authors approved the final version of the manuscript.
Conflicts of interest:The authors declare that there is no conflict of interest for the publication of this article.
Data availability statement:Not applicable.
Open access statement:This is an open access journal,and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License,which allows others to remix,tweak,and build upon the work non-commercially,as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- From the dust: extracellular vesicles as regulators of development and neuroregeneration
- Targeting epidermal growth factor receptor signaling to facilitate cortical injury repair?
- Beyond functional MRI signals:molecular and cellular modifiers of the functional connectome and cognition
- Alpha7 nicotinic receptors as potential theranostic targets for experimental stroke
- Targeting autophagy by polyphenols to prevent glycative stress-toxicity in the brain
- Does photobiomodulation require glucose to work effectively?