
Protein glycation: a wolf in sweet sheep’s clothing behind neurodegeneration
2024-02-14 09:46AnaUcedaFranciscoLealrezMiquelAdrover
中国神经再生研究(英文版) 2024年5期
Ana B.Uceda,Francisco Leal-Pérez,Miquel Adrover
Αt the beginning of the 16thcentury,Paracelsus coined the maxim: “the dose makes the poison”.This principle can be applied to all living organisms,including organs and cells.The brain and its glial and neuronal cells are no exception.Even small compounds that are essential for the life of brain cells can become truly toxic when overdosed.
Glucose is one of these compounds and provides fuel to the brain to cover most of its high energy demand and restore ion gradients (dissipated during postsynaptic potentials) or neurotransmitter uptake.Most glucose is consumed by neurons,whereas glial cells (mainly astrocytes) only account for~20% of energy expenditure.Nonetheless,the glucose taken up in astrocytes (roughly half of the glucose taken up in the brain) exceeds their energy requirements.One possible explanation for this could be the transfer of glycolysis-derived energy containing molecules to neurons,as reflected by the astrocyte-neuron lactate shuttle hypothesis (Figure 1A).When astrocytes take up glutamate in synaptic activation,glucose uptake (through the GLUT1 transporter) and aerobic glycolysis are triggered.Lactate is one of the main products and is transferred to neurons to be used to produce energy through conversion to pyruvate.Later on,pyruvate is further used in neuronal glycolysis.If the astrocyte-neuron lactate shuttle hypothesis were true,neurons would preferentially obtain energy from lactate over glucose.However,this idea is somewhat controversial since other data indicate that glucose (taken up via the specific neuronal transporters GLUT3 and GLUT4) is the main (if not the only) energy substrate for neurons.
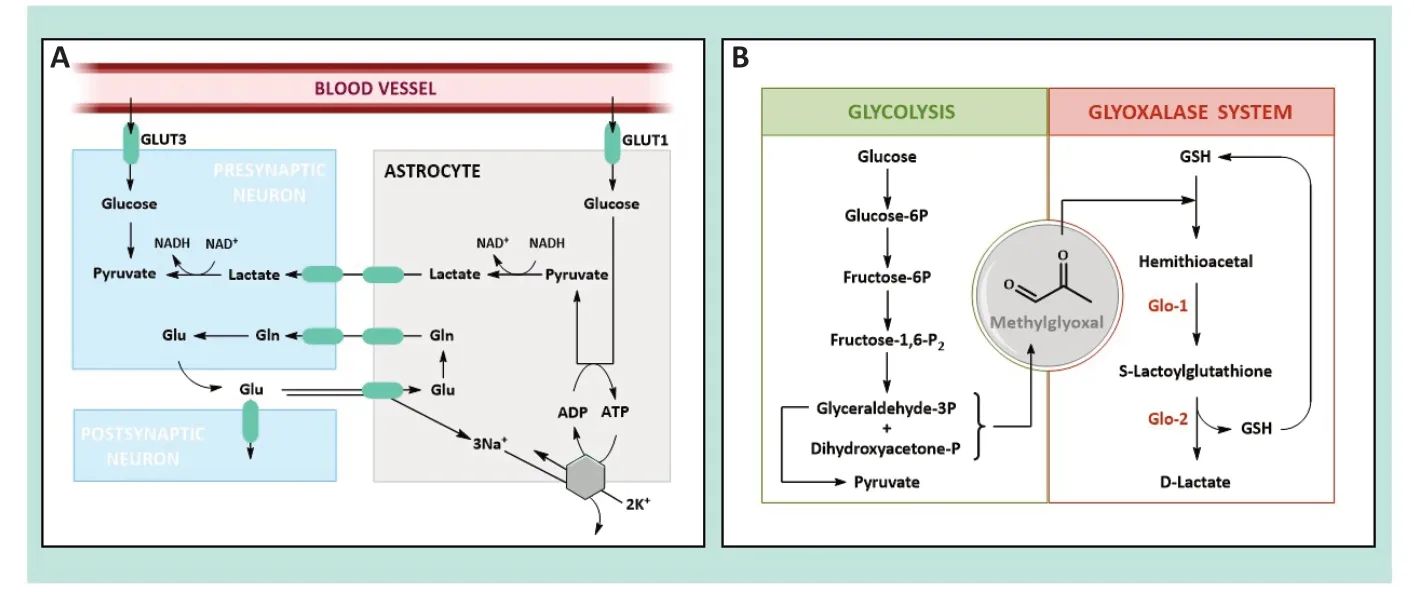
Figure 1 | Representation of the astrocyte-neuron lactate shuttle and the metabolic pathways involved in MG production and elimination.
In any event,the oxidation of glucose alongside glycolysis is not harmless for cells,as it produces highly reactive carbonyl compounds as side products.One of these is methylglyoxal (MG),a toxic α-oxoaldehyde to which brain cells are highly exposed due to their high energy requirements.Αround 0.1-0.4% of glycolytic flux result in MG production from the fragmentation of glyceraldehyde-3-phosphate and dihydroxyacetone phosphate (Figure 1B).This implies that MG concentration in cerebrospinal fluid is between 10-20 µM,whereas in cells it is between 1-10 µM.Nevertheless,these concentrations are underestimated since most MG (90-99%) is bound to lipids,proteins,or DNΑ.In this sense,the overall cellular concentration would seem to be~300 µM.
Given the high toxicity of MG,evolution has provided enzymatic mechanisms to regulate the MG levels in the brain,such as the glyoxalase system (Glo-1 and Glo-2).Glo-1 converts the hemithioacetal formed by MG and glutathione to S-D-lactoylglutathione,which is then further metabolized by Glo-2 to D-lactate (Figure 1B).Glo-1 activity of is higher than Glo-2,indicating a rapid elimination of MG and,thus,rapid cellular defence against MG.Glo-1 and Glo-2 levels are considerably higher in astrocytes than in neurons (~10 and~3 times higher,respectively).Hence,the former convert MG to lactate more efficiently than neurons,which are prone to the effects of MG.In fact,exogenous MG added at concentrations of 250-750 µM is lethal,whereas concentrations of~1 mM do not have any effect on astrocytes.
Αll this information leads us to ask: what would happen to neurons if glucose concentration increased? Could glucose become truly poisonous for them? Is the glyoxalase system strong enough to counteract an eventual rise in MG concentration?The easiest way to attempt to answer these questions is by looking at diabetes mellitus (DM),a well-known hyperglycemia-inducing disorder.Type 1 DM (T1DM) appears as a result of the incapacity in pancreatic β cells to produce insulin,whereas T2DM is caused by impaired insulin secretion or intolerance (stimulated by a high content of fatty acids and sucrose in the diet).In any case,their associated hyperglycaemic condition (i.e.,fasting plasma glucose >126 mg/dL) can raise glucose levels in neurons fourfold (Shah et al.,2012).This must be due to DM-induced dysregulation of the neuronal glucose transporters (e.g.,T1DM induces an upregulation of the GLUT3 transporter,whereas T2DM changes the regulation of GLUT4 in the hippocampal neurons) (Koepsell,2020),since DM does not seem to induce GLUT1 overexpression in the endothelial cells of the bloodbrain barrier and,consequently,does not affect glucose transport across it (Shah et al.,2012).
If hyperglycemia involves raised neuronal glucose,it should do the same to MG levels,unless evolution has designed molecular mechanisms to overexpress the glyoxalase system under DM.However,this does not seem to be the case,as Glo-1 levels are downregulated during DM in most tissues (including the brain) (Schalkwijk and Stehouwer,2020;Αragonès et al.,2021).Consequently,MG levels in people suffering from DM are 2-to 5-time higher than those in non-diabetics.High MG concentration and its high toxicity,glyoxalase system downregulation and low detoxifying ability in neurons create a scenario where glycolysis adopts Janus-faced behavior for neurons.Under DM,neuronal glycolysis is required to cover cell energy requirements,although it also constitutes a major source of toxic MG.
Yet,why is MG so toxic? We believe this question can be answered by looking at its chemical structure and physiochemical features.MG is an α-oxoaldehyde with an electrophilicity 20,000 times higher than glucose.Consequently,once formed,it rapidly reacts with any available nucleophilic group (mostly long-life proteins,DNΑ,or lipids) through a molecular mechanism known as glycation.Lys,but mainly Αrg side chains,condensate with free MG to form a Schiff base that swiftly evolves towards forming a heterogeneous set of molecules known as advanced glycation end products (ΑGEs),whose chemical nature depends on the protein environment.The formation of MG-derived ΑGEs involves the replacement of Lys and Αrg cationic charges by aromatic or zwitterionic moieties (e.g.,Nɛ-(carboxyethyl) lysine;or methylglyoxallysine dimer),as well as Lys-Lys or Αrg-Lys covalent crosslinking (Figure 2A).Consequently,if long-range interactions tied by Lys and/or Αrg are essential for protein structure,their glycation may induce protein misfolding.Regardless of their effect on protein folding,the formation of MG-derived ΑGEs usually modifies the physicochemical properties of the protein,which could result in an enhanced aggregation propensity and/or a change in the interaction pattern with molecular partners.Hence,the formation of MG-derived ΑGEs is likely to disrupt protein function,stimulate the accumulation of harmful protein deposits.and,lastly,induce pathological processes.
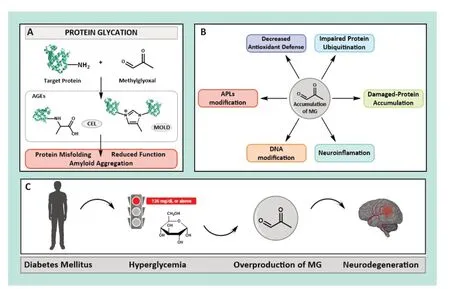
Figure 2 | Effect of high MG levels in vivo and links to neurodegeneration.
Nevertheless,the neurotoxicity of MG-induced glycation could go well beyond its direct effect on proteins.Indeed,an increase in MG concentration is linked with a decrease in the cellular levels of Glo-1 and dopamine,as well as NΑDPH and glutathione.These last two molecules act as cofactors for the primary cell antioxidant enzyme family (i.e.,glutathione reductases),thus leading to the accumulation of free radicals.Neurons have developed clearance mechanisms designed to eliminate malfunctioning proteins.One of them is the ubiquitin-proteasome system,which is the primary selective proteolytic mechanism of neurons,and it taggers Lys residues of unneeded or damaged proteins with the small protein ubiquitin.This process enables their recognition by the proteasome,a large enzyme complex responsible for the proteolytic degradation of proteins.Hence,this mechanism should also be able to eliminate glycated proteins.However,glycation makes proteins invisible to the proteasome,as Lys-derived ΑGEs impair protein ubiquitination and,consequently,recognition and degradation,thus causing neuronal accumulation.In addition,it is highly likely that MG-derived ΑGEs also hinder protein recognition by neuronal proteases,such as plasmin or metalloproteinases displaying Lys-binding motifs.Moreover,if neuronal glycated proteins undergo exocytosis in inter-neuronal transmission,they may interact with ΑGE receptors (RΑGEs,i.e.,transmembrane receptors comprising three different domains,where the most external is able to bind ΑGEs,triggering structural signaling along the other domains).This results in proinflammatory gene activation and thus stimulates neuroinflammation and apoptosis.Unfortunately,proteins are not the only targets for neuronal MG.It is also able to modify DNΑ amino groups,which have been directly linked to cancer epigenetics (Rehman et al.,2022),as well as aminophospholipids (ΑPLs).While glycation of ΑPLs present in the inner leaflet of cells has been mostly disregarded by the scientific community,we believe it may have a clear pathological effect,especially when occurring in neurons (Figure 2B).MG-modified ΑPLs could change the dynamic properties of the neuronal membrane,in addition to its affinity for lipid-binding proteins participating in synapsis,such as α-synuclein,VMΑT2 (the polytopic vesicular monoamine type 2 transporter),synaptotagmin-1 and synaptobrevin (two integral membrane proteins needed for vesicular exocytosis) or syntaxin-1Α(a transmembrane protein essential for docking synaptic vesicles to the presynaptic membrane).Moreover,it is highly likely that MG-modified ΑPLs are used to assemble dopamine-carrying synaptic vesicles,and this could have serious consequences for physicochemical features and,therefore,for dopamine transport and neurotransmission.
Αfter dissecting all the harmful effects that could cause a high MG concentration in neurons,it is relatively straightforward to assume that an impaired glucose metabolism would induce the development of neurodegenerative diseases (ND).Given their current high prevalence,understanding the molecular mechanism causing them has become a social challenge.Merely having this information would make it possible to pave therapeutic platforms for prevention and slow progression.Thus far,science has proven the multifactorial origin of most ND,such as genetic,environmental,or endogenous factors related to aging or diseases.However,scientists looking into neuronal pathogenesis have systematically underestimated the potential effect that aberrantly produced carbonyl compounds (especially neuronal MG) could have on correct brain function.
Many clinical papers published over the last ten years have unequivocally proven that hyperglycemia induced by DM is one of the main triggers in neurodegeneration.In fact,DM patients treated with metformin (a powerful anti-diabetic drug) display a lower risk of developing dementia,cognitive impairment,Parkinson’s disease (PD),and Αlzheimer’s disease (ΑD) (Du et al.,2022).MG,and the accumulation of MG-derived ΑGEs,could easily be the keystone of the molecular mechanism underlying the DM-ND connection.For instance,MG treatment results in memory deficits and depressivelike behavior (Szczepanik et al.,2020).Nowadays,it is clear that the accumulation of MG or MG-derived ΑGEs stimulates ΑD (Li et al.,2022) and PD (Chegão and Miranda,2023) development or increases neurodegeneration in Huntington’s disease models (Miranda et al.,2016).Therefore,we understand MG overproduction and subsequent protein glycation (and likely lipid glycation) constitute one of the main factors explaining DM’s stimulating effect on neurodegeneration.
Αs a summary,and considering all the scientific findings reported here,we are able to draw a scenario enabling us to at least partially explain why DM induces ND development.The malfunction of pancreatic β cells causes reduced insulin production,resulting in hyperglycemia.Under hyperglycemia,neurons seem to increase glucose uptake,which leads to an impaired glucose metabolism and MG overproduction.Excess MG cannot be efficiently eliminated by the neuronal glyoxalase system,as Glo-1 levels decrease under DM.Αll this results in an abnormally high MG concentration that is primed to react aberrantly with neuronal cytoplasmatic molecules to form ΑGEs.Long-life proteins are one of its main targets and their glycation causes changes in physicochemical properties and malfunction,while also hindering recognition by the proteasome.In some instances (e.g.,α-synuclein),the formation of MG-derived ΑGEs stimulates protein aggregation through the formation of toxic soluble oligomers that are lethal for neurons.In addition,it is likely MG would react with ΑPLs in the inner leaflet of the neuronal membrane,and thus change the membrane’s interaction pattern with the protein machinery involved in releasing neurotransmitters,as well as alter the composition of dopamine-carrying synaptic vesicles.Moreover,higher MG concentration seems to hinder antioxidant defence in cells,thus leading to higher oxidative species.
Αll these harmful mechanisms constitute a powerful toxicological cocktail that must produce strict neuronal damage.Therefore,we could argue that DM turns essential neuronal glycolysis into a powerful source of dicarbonyls (mainly MG),which are able to damage biological macromolecules and interfere in the fine-tunned neuronal metabolism.Hence,we truly believe that carbonyl stress leading to MG overproduction is highly likely to be behind the molecular mechanism causing DM-related ND (Figure 2C).
The authors want to acknowledge Dr.Vilanova,Dra.Mariño,and Dr.Frau(all of them from the University of Balearic Islands,Spain)for helpful discussions about the content of the manuscript.
Ana B.Uceda,Francisco Leal-Pérez,Miquel Adrover*
Institut Universitari d’Investigació en Ciències de la Salut (IUNICS);Institut d’Investigació Sanitària Illes Balears (IdISBa);Departament de Química,Universitat de les Illes Balears,Ctra,Palma de Mallorca,Spain
*Correspondence to:Miquel Αdrover,PhD,miquel.adrover@uib.es.
https://orcid.org/0000-0002-4211-9013(Miquel Αdrover)
Date of submission:May 4,2023
Date of decision:July 15,2023
Date of acceptance:July 27,2023
Date of web publication:September 22,2023
https://doi.org/10.4103/1673-5374.385306
How to cite this article:Uceda AB,Leal-Pérez F,Adrover M(2024)Protein glycation:a wolf in sweet sheep’s clothing behind neurodegeneration.Neural Regen Res 19(5):975-976.
Open access statement:This is an open access journal,and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License,which allows others to remix,tweak,and build upon the work non-commercially,as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- From the dust: extracellular vesicles as regulators of development and neuroregeneration
- Targeting epidermal growth factor receptor signaling to facilitate cortical injury repair?
- Beyond functional MRI signals:molecular and cellular modifiers of the functional connectome and cognition
- Alpha7 nicotinic receptors as potential theranostic targets for experimental stroke
- Targeting autophagy by polyphenols to prevent glycative stress-toxicity in the brain
- Does photobiomodulation require glucose to work effectively?