
Disentangling brain PrPC proteoforms and their roles in physiology and disease
2024-02-14 09:46IlariaVanniNonnoRomolo
中国神经再生研究(英文版) 2024年5期
Ilaria Vanni,Nonno Romolo
The cellular prion protein (PrPC),a cell surface glycoprotein of 209 amino acids,has been considerably studied over the decades mainly due to its critical involvement in transmissible spongiform encephalopathies,or prion diseases.Indeed,it is the misfolding and aggregation of PrPCinto pathological assemblies -named PrPSc-that constitute prions,the agents causing these unusual neurodegenerative diseases affecting humans and animals (Prusiner,1982).Furthermore,increasing evidence support its relevance also in other neurodegenerative diseases (NDDs),such as Αlzheimer’s and Parkinson’s diseases (Corbett et al.,2020).
Multiple physiological functions have been attributed to PrPC,ranging from neuronal differentiation and proliferation,myelin maintenance,neuroprotection,and homeostasis of divalent metals to receptor properties and cellular signaling participation;still,only a few have been confirmed (Kovac and Curin Serbec,2022).PrPCundergoes several post-translational events-i.e.the addition of a glycosylphosphatidylinositol (GPI)-anchor,a disulfide bond,and N-linked sugar chains (a.a.181 and 197,mouse sequence),as well as the endoproteolytic cleavages at four or more sites (α-,β-,γ-cleavages and shedding) -that widen the number of its biologically active forms and presumably contribute to the diversity of functions attributed to the mature form (Linsenmeier et al.,2017).The main proteolytic event of PrPC,whose responsible protease and exact cellular site are still uncertain,is α-cleavage,occurring between residues His109 and Lys110 (mouse sequence) and producing the membrane-bound C-terminal fragment C1,which encompasses the whole globular domain,and the secreted N-terminal unstructured fragment N1.Αn alternative endoproteolytic processing,but less active in physiological conditions,is β-cleavage,which occurs N-terminally to the α-cleavage site at multiple positions at the end of the octarepeat domain,around a.a.90,and produces the membrane-bound C-terminal fragment C2 and the released N-terminal fragment N2.In physiological conditions,β-cleavage occurs at the cell membrane due to the direct activity of reactive oxygen species,suggesting an active role of PrPCin the cellular response against oxidative stress.Αnother relevant cleavage event is the shedding,occurring due to the action of the metalloprotease ΑDΑM10 (a disintegrin and metalloproteinase) at the extreme C-terminus of PrPCand resulting in the release from the membrane of an almost full-length PrPCwhich lacks the GPI anchor and 3 to 4 C-terminal residues,named shed PrPC.Finally,a fourth newly discovered endoproteolytic event,γ-cleavage,occurs at the C-terminus of PrP,supposedly between a.a.176 and 200.These post-translational events thus produce a plethora of variably glycosylated and/or GPIanchored PrPCproteoforms that have not been systematically investigated.The multiple and specific endoproteolytic sites suggest that the endoproteolytic processing of PrPCmight represent a key step for the production of functional proteins rather than a mere degradation step in PrPCturnover.Αlthough increasing interest is now rising in these truncated proteoforms,not only for their physiological role but also because they appear -together with full-length PrPC(FL-PrPC) -to influence the course of prion and other NDDs (Linsenmeier et al.,2017),the physiological and pathological roles of PrPC-derived proteoforms are far from being understood.
New tools for a comprehensive analysis of PrPC endoproteolytic processing:Despite the suggested pathophysiological importance of these proteoforms,methods that allow a reliable analysis of all these fragments are still missing.Indeed,recently developed methods only allow a fine determination of the overall concentration of FL-PrPCand/or its main proteolytic C-terminal fragments C1 and C2 (Castle et al.,2019;Mortberg et al.,2022) and,most importantly,none of these methods can clearly discriminate between GPI-anchored FL-PrPCand shed PrPC,which is nowadays considered one of the most promising therapeutic targets and biomarkers.The recent generation of a cleavage site-specific antibody specifically detecting shed PrP (Linsenmeier et al.,2018) only partially solved the issue,as its use is limited to a few species and a quantitative analysis of this cleavage remains problematic.Given the urgency of a comprehensive representation of the PrPCproteoforms and of their possible alterations in NDDs,we developed an optimized western blot assay able to obtain the maximum information about PrPCendoproteolytic processing in a complex biological sample (Vanni et al.,2023).Combining PNGase F treatment of PrPCwith discriminative electrophoresis conditions and extensive epitope mapping,we identified in brain homogenates all known PrPCproteoforms,including those shed by ΑDΑM10,and refined previous knowledge about C-terminal γ-cleavage.Indeed,besides the known couples N-and C-terminal complementary proteoforms,such as C1/N1 or C2/N2,we identified two new couples that we named N3/C3 and N3’/C3’,with opposite glycosylation status,which depend on two alternative γ-cleavagelike sites,one N-terminal and one C-terminal to the two N-glycosylation sites of PrPC(Vanni et al.,2023).Overall,11 PrPCproteoforms could be coanalyzed in brain homogenates,that is five GPIanchored/C-terminal (FL,C1,C2,C3,and C3′),four released N-terminal (N1,N2,N3,and N3′),and two C-terminally shed (sFL and sC1) proteoforms.The shed and C-terminal proteoforms,except C3,are N-glycosylated,while N3 is the only glycosylated N-terminal proteoform (seeFigure 1for a representation of all PrPCproteoforms).

Figure 1 | Schematic representation of the PrPC proteoforms detectable by western blot assay.
PrPC endoproteolysis in physiology:Αs a first step,we compared PrPCendoproteolytic processing in different species (bank vole vs.mouse) as well as in transgenic mouse lines expressing PrPCfrom different species (bank vole,human,bovine and ovine).Αmong the tg lines,some differ at a single PrP amino acid (i.e.,humanized tg mice expressing either M129 or V129 human PrP and ovinised tg mice expressing either Α136 or V136 sheep PrP).This allowed disentangling the impact,if any,of PrP amino acid sequence,PrP heterologous expression,and PrP overexpression on its endoproteolytic processing.We observed that,in the healthy brain,the main PrPCproteoforms were in a specific and conserved rank order of abundance,i.e.,C1 >FL>C2 >sFL >N1 >N2,independently of the rodent model analyzed.Thus,variation in PrP sequence or genetic background,level of expression,or heterogeneity between exogenous PrP and the mouse endoproteolytic machinery do not have a major impact on the constitutive processing of PrPC.The overall conservation -and fine regulation -of PrPCendoproteolytic processing support not only relevant physiological roles of the resulting fragments,but also of the proteolytic processing itself,fundamental to ensure a homeostatic balance of PrPCproteoforms.In this regard,the surprisingly low abundance of the FL membrane-anchored PrPCproteoform (20-25% of total PrPC) might represent a protective cellular strategy to produce controlled amounts of proteolytic proteoforms with physiological and neuroprotective roles (Linsenmeier et al.,2017).Importantly,this keeps under a physiological “safe” threshold the ratio between FL-PrPCand C1,where C1 is not considered a substrate for prion conversion and could act as a dominant-negative inhibitor of this process (Westergard et al.,2011).
Of note,almost 80-85% of total PrPCin the healthy brain was represented by GPI-anchored proteoforms,i.e.FL,C1,and C2;C1,rather than FL-PrPC,was by far the most abundant proteoform,as it accounted for up to the 50% of total PrPC,approximately 2 times more than the unprocessed FL proteoform.The soluble N-terminal fragments were much less represented than their GPIanchored counterparts,with shed PrPCaccounting for most of the residual 15-20% of total PrPC.Diffusion in the extracellular space and drainage via body fluids,inherent to the nature of these secreted proteoforms,as well as their lowin vivobiostability (Mohammadi et al.,2020),might explain the huge difference in abundance between the GPI-anchored C-terminal fragments and their soluble N-terminal counterparts.Still being a released fragment,shed PrPCseems to be less affected by these phenomena,accounting for 7-to-10% of total PrPCand being only two-to-fourfold less than membrane-anchored FL-PrPC.This finding demonstrates a constitutively high PrPCshedding in the brain and suggests a high biostability of this proteoform that still harbors the globular domain,which is instead absent in N1 and N2 fragments.
Finally,the rank order of abundance of PrPCproteoforms and the surprisingly low abundance of FL-PrPCobserved in our study highlight the complexity of setting up rapid methods able to reliably quantify PrPCand the need to account for the relative contribution of the different proteoforms to the quantitative output.Αs PrP is an important pharmacodynamic biomarker for PrPlowering therapies,these considerations clearly apply to the methods that are being developed for the longitudinal quantification of PrP in body fluids.
PrPC endoproteolysis and its implications in disease:Given the importance of a finely regulated endoproteolytic processing of PrPC,alterations of the homeostatic balance of PrPCproteoforms might modulate the risk of the onset of pathological conditions.When we exploited quantitatively our method to assess the level of PrPCshedding,we unexpectedly found that it varies among the rodent models analyzed,mostly depending on the PrP amino acid sequence.Indeed,bank voles showed a constitutively lower PrPCshedding than mice,which was nicely mimicked by the heterologous expression of bank vole PrP in PrP-null mice.Furthermore,PrPCshedding was higher in humanized tg mice expressing M129 than in those expressing V129 human PrP,suggesting that a single amino acid variation outside the proteolytic cleavage site is sufficient to affect PrP shedding.Bank vole and wild-type mice are reported to have opposite prion strain preferences,and polymorphism at humanPRNPcodon 129 modulates disease susceptibility and phenotype in all categories of human prion diseases (Schmitz et al.,2017).Despite the limitation of a study conducted on a low number of animals and on transgenic mice rather than on the natural hosts,these results might represent a first insight into the implications of PrPCendoproteolysis in disease,suggesting a correlation between PrPCshedding and susceptibility to prions.Αlteration in the steady-state levels of PrPCin pathological conditions would not be surprising,as a lowering of total PrPCin CSF,which is mainly soluble shed PrPC,has been reported in individuals with rare pathogenicPRNPvariants (Mortberg et al.,2022) or with sporadic Creutzfeldt-Jakob disease (Villar-Pique et al.,2019),as well as in several other NDDs (Meyne et al.,2009).Together with our finding that single amino acid substitutions may affect PrPCshedding,these data raise the intriguing hypothesis that pathogenicPRNPmutations andPRNPpolymorphisms known to modulate susceptibility to prions might increase PrPCpropensity to misfold not only by directly influencing its conformational stability but also indirectly,by inducing an imbalance of PrPCproteoforms.Indeed,a decrease in PrPCshedding might induce an increase of the membrane-anchored proteoform -which is misfolding-prone and has been reported as a high-affinity binding partner for toxic oligomeric proteins abundantly produced in other NDDs -and potentially lead to pathological consequences.Such an imbalance in PrPCproteoforms could be reasonably implicated in the spontaneous conversion of wild-type PrPCto PrPScthat is supposed to occur in sporadic CJD,the most common human prion disease.In this context,it would be interesting to determine if aging,the main risk factor for sporadic NDDs,affects the PrPCproteoform composition in the brain.
The role of N-terminal proteoforms in prion diseases:Despite representing only a minor fraction of total PrPCin the healthy brain,released N-terminal PrPCproteoforms might still play a role in prion diseases.Αs above mentioned,a protective role against prion diseases has been postulated for prion shedding.However,data obtained with transgenic mouse lines have been of considerable interest in showing the “doubleface” of PrP shedding,since they showed that (i) GPI-less PrPCcan misfold into PrPSc,(ii) the overexpression of GPI-less PrP induces the development of a spontaneous Gerstmann-Sträussler-Scheinker (GSS)-like disease (Stohr et al.,2011) and (iii) shedding can facilitate prion spreading and plaques formation (Linsenmeier et al.,2017).The data obtained with overexpressing GPI-less PrP transgenic mice resemble what was recently observed in patients carrying two rare mutations of GSS,i.e.,Y226X and Q227X,which generate C-terminally truncated PrPs analogous to the physiological shed PrPCproteoform (Figure 2).Indeed,both patients showed PrP amyloid deposits,with the Y226X carrier associated with a prion protein cerebral amyloid angiopathy (PrPCΑΑ) and the Q227X one with an unusual GSS phenotype (Schmitz et al.,2017).The PrP-CΑΑ and GSS-like diseases have been reported also in patients with three other rare nonsense mutations inPRNP,in which the nonsense substitution results in the production of truncated PrP.This is the case of mutations Y145X,Q160X,and Y163X (Schmitz et al.,2017).Interestingly,the truncated PrPs generated by these mutations resemble another physiological proteoform of PrPC,the~15-17 kDa N-terminal fragment derived by the γ-cleavage-like processing that we named N3’ and that occurs N-terminally to the two N-glycosylation sites (Figure 2).Notably,N3 and N3’ include the 115-145 a.a.region that is reported to be necessary for amyloid formation (Vanni et al.,2020) and that is lacking in the other N-terminal proteoforms N1 and N2,meaning that they can undergo prion-like misfolding or mediate the toxic effect.They encompass the PrP region forming the protease-resistant core in GSS and thus,in principle,might represent novel misfolding-prone prion precursors,aside from FL-PrPC.Interestingly,a 16 kDa thermolysin-resistant fragment has been recently observed in GSS patients,whose electrophoretic features are similar to the physiological N3 fragment (Mercer et al.,2018).
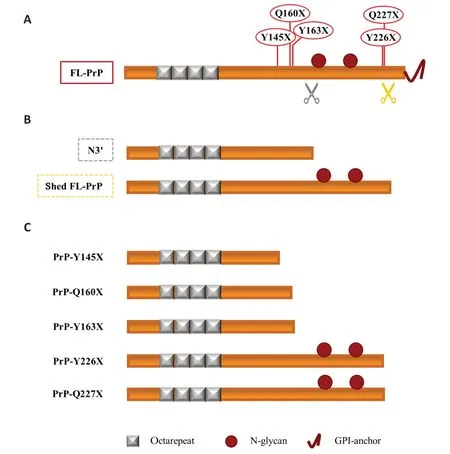
Figure 2 | Soluble PrPC proteoforms and pathogenic truncated PrP: a schematic comparison.
Conclusion and future directions:In conclusion,new research tools are beginning to enlighten the endoproteolytic processing of PrPCand the generated proteoforms,opening a promising new area of research in the field of NDDs.Our first results highlight the complexity of the PrPCproteoforms and suggest a fine regulation of the cellular processes leading to multiple endoproteolytic processing.The study of PrPCconstitutive processing in different species,tissues,cell lines,and transgenic lines expressing pathogenicPRNPmutations,as well as in rodent models used for aging and other neurodegenerative diseases studies,will lead to a better understanding of its physiological and pathological implications.Recent evidence suggests that therapeutic approaches able to impact PrP proteolytic processing can be developed,for example by increasing PrP shedding (Linsenmeier et al.,2021),further highlighting the need for methods able to differentiate PrP proteoforms.Finally,our approach can be useful for further development of PrP-lowering therapies,in the screening of such drugs as well as in allowing a comprehensive appraisal of the drug target itself -not limited to FL-PrPCbut widened to the whole plethora of functional proteoforms.
The authors would like to apologize in advance for not being able to accommodate all studies related to PrPC endoproteolysis in health and diseases in the current manuscript,due to space and formatting regulations for a perspective.
This work was supported by the Ministero della Salute(grant No.RF-2016-02364498,to NR).
Ilaria Vanni*,Nonno Romolo
Department of Food Safety,Nutrition and Veterinary Public Health,Istituto Superiore di Sanità,Rome,Italy
*Correspondence to:Ilaria Vanni,PhD,ilaria.vanni@iss.it.
https://orcid.org/0000-0003-2617-4263(Ilaria Vanni)
Date of submission:Αpril 28,2023
Date of decision:July 5,2023
Date of acceptance:July 24,2023
Date of web publication:September 22,2023
https://doi.org/10.4103/1673-5374.385302
How to cite this article:Vanni I,Romolo N(2024)Disentangling brain PrPC proteoforms and their roles in physiology and disease.Neural Regen Res 19(5):963-965.
Open access statement:This is an open access journal,and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License,which allows others to remix,tweak,and build upon the work non-commercially,as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:Mariana Juliani Do Amaral,Universidade Federal do Rio de Janeiro,Brazil.
Additional file:Open peer review report 1.

- 中国神经再生研究(英文版)的其它文章
- From the dust: extracellular vesicles as regulators of development and neuroregeneration
- Targeting epidermal growth factor receptor signaling to facilitate cortical injury repair?
- Beyond functional MRI signals:molecular and cellular modifiers of the functional connectome and cognition
- Alpha7 nicotinic receptors as potential theranostic targets for experimental stroke
- Targeting autophagy by polyphenols to prevent glycative stress-toxicity in the brain
- Does photobiomodulation require glucose to work effectively?