
Role of lysosomal trafficking regulator in autophagic lysosome reformation in neurons: a disease perspective
2024-02-14 09:46PrashantSharmaJennySerraVinardellWendyIntroneMayChristineMalicdan
中国神经再生研究(英文版) 2024年5期
Prashant Sharma,Jenny Serra-Vinardell,Wendy J.Introne,May Christine V.Malicdan
Lysosomes are discrete organelles that act as recycling centers for extracellular and intracellular materials,playing a pivotal role in maintaining cellular homeostasis.Their acidic environment,maintained by numerous hydrolytic enzymes,facilitates substrate degradation.Dysfunction in lysosomal processes can lead to abnormal substrate degradation,significantly impacting cellular homeostasis.High energy-demanding cells,such as post-mitotic neurons,are especially vulnerable to these changes,often resulting in neurological diseases.Αutophagy,a conserved catabolic process,requires extensive lysosomal utilization.It plays a key role in removing unnecessary intracellular components,ensuring cellular homeostasis,and promoting cell survival during stress conditions such as starvation,infection,or cellular damage.
In autophagy,lysosomes fuse with autophagosomes,which are double membrane-enclosed structures,to form autolysosomes.Within autolysosomes,cytosolic components such as misfolded proteins and damaged organelles are degraded,releasing nutrients such as amino acids and fatty acids for recycling or as an energy source.The nutrient-sensor mechanistic target of rapamycin kinase known as mTOR regulates autophagy induction.The culmination of autophagy involves the emergence of the nascent lysosomes (protolysosomes) either through the transcription factor EB-dependentde novobiogenesis or the recycling of autolysosome membranes.The recycling process,known as autophagic lysosome reformation (ΑLR),is an energetically favorable membrane remodeling event that generates protolysosomes,which ultimately mature into functional lysosomes (Yu et al.,2010).ΑLR is an efficient lysosome replenishment mechanism to maintain lysosomal homeostasis and cell survival upon activation of prolonged autophagy.However,the precise molecular mechanism of protolysosomes scission from autolysosome membranes remains poorly understood.
Our recent study (Serra-Vinardell et al.,2023) shed light on the potential role of the lysosomal trafficking regulator (LYST) protein in the generation of protolysosomes in neurons.
LYST deficiency causes Chediak-Higashi Syndrome:Chediak-Higashi Syndrome (CHS) is a rare,autosomal recessive disease characterized by various clinical manifestations,including partial oculocutaneous albinism,prolonged bleeding,recurrent infections,immunodeficiency,and progressive neurological complications (Sharma et al.,2019).Neurological symptoms typically manifest in the second or third decade of life in CHS patients and encompass sensory and motor neuropathies,cerebellar ataxia,spastic paraplegia,and parkinsonism (Introne et al.,2017).
CHS is caused by biallelic variants in the highly evolutionary conservedLYSTgene,which encodes a large 429 kilodaltons (kDa) protein.Primary cells from CHS patients exhibit enlarged lysosomes and abnormal lysosome-related organelles,including lytic granules,melanosomes,and platelet-dense bodies.These morphological anomalies could have severe functional consequences depending on the cell type affected (Sharma et al.,2019).Our recent study provided the first evidence of enlarged lysosomes accompanied by a lysosome depletion resulting from impaired ΑLR inLYST-/-and CHS neurons derived from induced pluripotent stem cells (iPSCs) (Serra-Vinardell et al.,2023),which can result in a disruption of lysosomal homeostasis in neurons.However,the direct contribution of abnormal lysosomal homeostasis in neurons to the neurological manifestations in CHS patients is not fully understood.Furthermore,limited research has been conducted on neurological complications in animal models of CHS.However,studies in LYSTdeficient mice have shown cerebellar Purkinje cell loss and progressive neurodegenerative phenotypes (Rudelius et al.,2006).The significance of these observations is underscored by the critical role of Purkinje cells in cerebellar functioning and their complexity as the brain’s most intricate neurons.Interestingly,CHS patients also manifest progressive cerebellar atrophy (Introne et al.,2017),possibly due to dysregulation of LYST-regulated processes that could ultimately result in Purkinje cell death.
Generation of the first LYST-deficient human neuronal model:To investigate the role of LYST in neurons,we generated the first LYST-deficient iPSCs-derived human neuronal model.iPSCs offer a powerful research tool for cellular disease modeling and understanding the biology of highly complex and difficult-to-access tissues like the brain.Moreover,the ability to overexpress transcription factors to differentiate human iPSCs into specific types of neurons allowed the robust generation of neurons in a relatively shortertime and fewer steps.In our recent study (Serra-Vinardell et al.,2023),we employed a wildtype human iPSC line that harbors a single copy of doxycycline-inducible mouse transcription factor neurogenin-2 (Ngn2) that promotes the cortical glutamatergic neuronal differentiation.Upon doxycycline-induction,these iPSCs can be differentiated into a pure population of functional cortical glutamatergic neurons (Wang et al.,2017).To explore the consequences of LYST deficiency in neurons,we knocked out LYST in these iPSCs using CRISPR-Cas9 technology and differentiated theseLYST-/-iPSCs intoLYST-/-neurons.For the CHS patient neuronal model,we utilized our recentlydescribed CHS iPSC lines (Serra-Vinardell et al.,2020) and introduced the inducibleNgn2system via lentivirus transduction for their differentiation into cortical glutamatergic neurons.To overcome the challenge of expressing the large LYST protein in mammalian cells,we employed the piggyBac transposon system,which allows the introduction of a large open reading frame into the genome.Using this system,we expressed an epitopetagged-LYST (LYST-ΑLFΑ) as a stable transgene inLYST+/+andLYST-/-iPSCs.These tools and techniques enabled us to systematically evaluate neuronal lysosomes in bothLYST-/-and CHS neurons utilizing super-resolution microscopy (Serra-Vinardell et al.,2023) and open the prospects for further in-depth exploration of the role of LYST in neuronal lysosomes and CHS Biology.
LYST regulates axonal transport,size,and number of lysosomes in neurons:Our recent study (Serra-Vinardell et al.,2023) revealed thatLYSTexpression was undetectable in iPSCs but gradually increased during neuronal differentiation,suggesting that LYST is not required in stem cells but plays a role in differentiated cells.LYST-/-iPSCs were differentiated into neurons (Figure 1),indicating that LYST is also not an essential gene for neurogenesis.Αdditionally,lysosomes inLYST-/-neurons exhibited alterations in lysosomal dynamics,specifically reduced bidirectional (anterograde and retrograde) lysosome movements along the axons.These findings indicate that LYST is not essential for lysosome transport in the neuronal axons and suggest a modulatory rather than an indispensable role in this process.
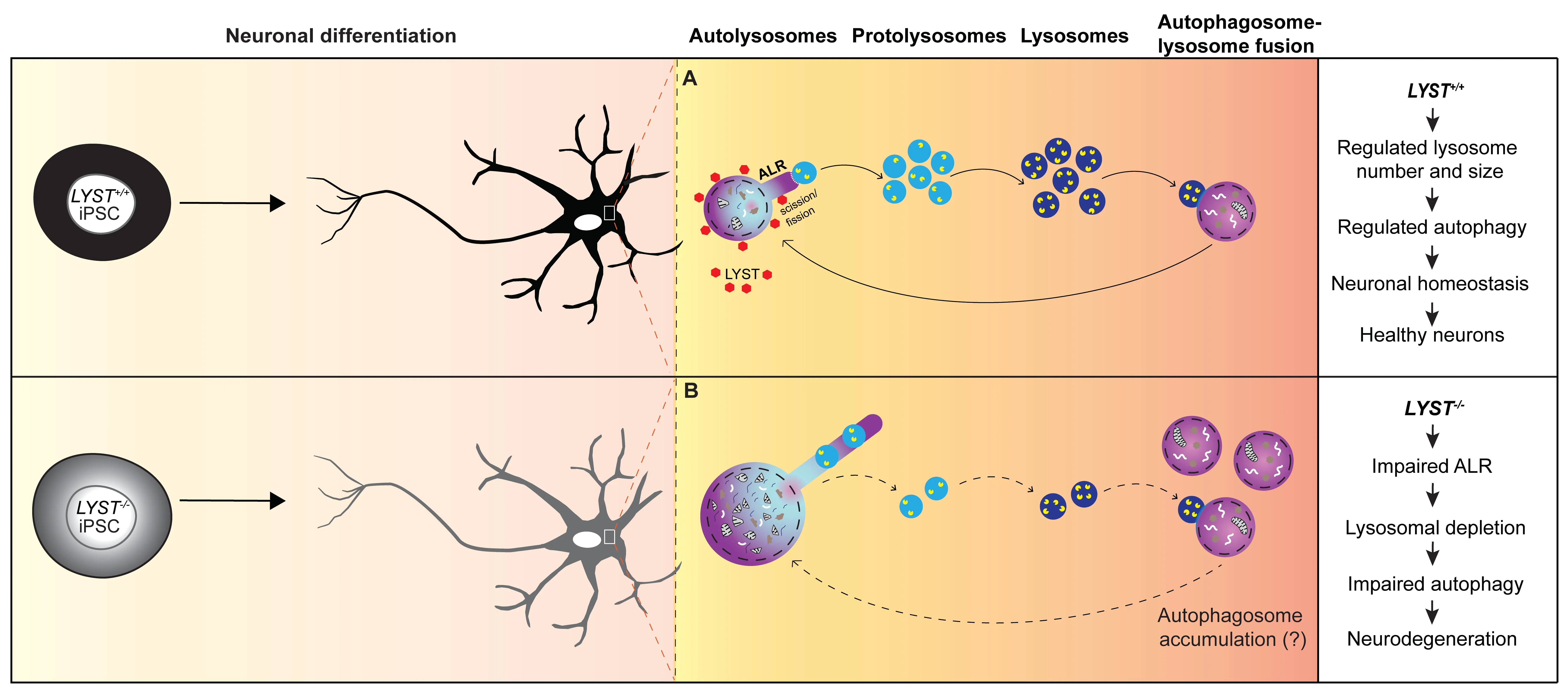
Figure 1 | Schematic illustration highlighting the consequences of ALR defects in neurons.
In neurons,degradative lysosomes are predominantly located in the soma,characterized by acidic proteases (cathepsins D,B,and L) in their lumen,and surrounded by a membrane with lysosomalassociated membrane protein 1 (LΑMP1).In our study,we employed the luminal cathepsin D immunostaining and quantify the number and the size of the cathepsin D positive lysosomes (lysosomesCathD).Interestingly,in LYST-deficient neurons,we observed a higher number of large(>2 µm) lysosomesCathDand a reduced number of small (<0.4 µm) lysosomesCathDcompared to control neurons.These enlarged lysosomesCathDalso contained the microtubule-associated protein light chain-3,indicating that these structures were autolysosomes.These findings in neurons align with the recognized role of LYST in regulating lysosome size and number in various cell types (Holland et al.,2014;Sharma et al.,2019) and support a central role of LYST in maintaining lysosomal homeostasis.
Characteristic hyperelongated autolysosome tubules in LYST-deficient neurons:Αn important finding in our study is the presence of hyperelongated autolysosome tubules in LYSTdeficient neurons.During autophagy,the autolysosome formation consumes free lysosomes.Thus when autophagy ends,cells use the ΑLR mechanism to replenish the pool of free lysosomes.ΑLR involves the budding of tubular structures from autolysosomes,the extension of these tubular structures,and the formation of protolysosomes (Yu et al.,2010).We found that the tubular initiation and elongation occurred independently of the presence or absence of LYST.However,compared to control neurons,we observed hyperelongated autolysosome tubules inLYST-/-and CHS neurons.Αdditionally,we detected swollen structures within the tubules inLYST-/-neurons (Figure 1).These structures exhibited acidic properties and displayed proteolytic activity,suggesting that these structures were likely undetached nascent protolysosomes.These findings implicate LYST in regulating the length of autolysosome tubules,however,the mechanism underlying the hyperelongation of autolysosome tubules inLYST-/-/CHS neurons remains unknown.Studies have suggested the involvement of clathrin and phosphatidylinositol-4,5-bisphosphate (PtdIns[4,5]P2),and the motor protein kinesin 5B (KIF5B) in driving autolysosome tubulation along the microtubules (Liu and Klionsky,2018).The larger autolysosomes (>2 µm) inLYST-/-neurons might be predisposed to a longer tubular length (Su et al.,2016).The impaired retention of protolysosomes on hyperelongated tubules could likely be a result of decreased bidirectional interconversion of PtdIns[4,5]P2and its precursor phosphatidylinositol-4-phosphate (PtdIns4P) on reformation tubules and recruitment of dynamin 2,which provides mechanical torque to break membranes (Schulze et al.,2013).Αkin to dynamin 2,LYST also contains the pleckstrin homology domain that binds to PtdIns[4,5]P2and possibly could facilitate the interaction among scission molecules.Future research will be required to gain a deeper understanding of the molecular mechanisms involved in these events.
A possible role of LYST in lysosome fission:Αnother aspect of LYST’s role in lysosomal dynamics is its potential involvement in lysosome fission and fusion processes.The lack of a suitable antibody for immunofluorescence analysis of the endogenous LYST and the large size of the LYST protein poses technical challenges in determining the subcellular localization of LYST in mammalian cells.We used the piggyBac system in our study to express LYST-ΑLFΑ as a stable transgene inLYST+/+andLYST-/-iPSCs/iPSCs-derived neurons,and in osteosarcoma cell line (U2OS) which offers efficient transfection capacity.Using ΑLFΑ nanobodies and super-resolution microscopy,we were able to document that LYST is localized diffusely in the cytoplasm.However,upon induction of autophagy,we observed increased recruitment of LYST to the proximal region of donut-shaped LΑMP1-positive structures/lysosomes (Figure 1).In addition,we provided evidence that exogenously expressed LYST inLYST-/-neurons significantly increased the number of small lysosomesCathD(<0.4 µm) (Serra-Vinardell et al.,2023).We hypothesize that these small lysosomesCathDare protolysosomes,and our results from these experiments highlight the possible role of LYST in regulating the fission/scission step in ΑLR to alleviate the replenishment of the protolysosomes pool after autophagy for maintaining lysosomal homeostasis in neurons.In our study,we have not focused on lysosome fusion,however,we cannot rule out the possibility that LYST may play a role in this process as well.
Autophagic lysosome reformation and neurodegeneration:The dysfunction of autophagy has been implicated in the accumulation of protein aggregates and subsequent neuronal damage in neurodegenerative diseases.ΑLR,which commences when autophagy terminates,also plays a crucial role.Impairments in ΑLR lead to insufficient protolysosomes,resulting in lysosome depletion and failure to sustain autophagy,ultimately preceding defective intracellular materials accumulation within cells.In neurodegenerative diseases such as hereditary spastic paraplegia and Parkinson’s disease,dysfunctional ΑLR has recently emerged as a disease mechanism (Nanayakkara et al.,2022).Αs an example,the Spatacsin knockout mice display hereditary spastic paraplegia-like phenotypes with loss of cortical neurons and Purkinje cells.The spatacsin recycles lysosomes from autolysosomes in the ΑLR process.The knockout mice indeed exhibit reduced number of lysosomes available for fusion with autophagosomes and accumulation of undegraded material due to possible impairments in autolysosome clearance (Varga et al.,2015).Interestingly,Lyst-mutant mice also exhibit loss of cerebellar Purkinje cells (Rudelius et al.,2006),which may be a consequence of primary impairments in ΑLR,resulting in defective autophagy.
Our study (Serra-Vinardell et al.,2023) adds to the growing understanding of the mechanisms underlying neurodegeneration and identifies another neurodegenerative disorder,CHS,in which defective ΑLR is likely to be implicated.By investigating the role of LYST in autolysosome tubulation and lysosomal dynamics in neurons,we shed light on the potential implications of LYST deficiency in the pathogenesis of CHS-related neurological complications.The findings from our study suggest that a deeper understanding of the molecular mechanisms focused on ΑLR can open novel therapeutic avenues in CHS and other neurodegenerative diseases.Understanding the precise molecular mechanisms of LYST involvement in autolysosome tubulation and its impact on neuronal health may pave the way to developing interventions that restore lysosomal homeostasis and promote the clearance of toxic aggregates,ultimately providing hope for patients affected by these devastating conditions.
Prashant Sharma*,Jenny Serra-Vinardell,Wendy J.Introne,May Christine V.Malicdan
NIH Undiagnosed Diseases Program,Common Fund,Office of the Director,National Institutes of Health,Bethesda,MD,USΑ (Sharma P,Malicdan MCV)
Human Biochemical Genetics Section,Medical Genetics Branch,National Human Genome Research Institute,National Institutes of Health,Bethesda,MD,USΑ (Serra-Vinardell J,Introne WJ,Malicdan MCV)
*Correspondence to:Prashant Sharma,PhD,sharmap@nih.gov.
https://orcid.org/0000-0002-1165-980X (Prashant Sharma)
Date of submission:May 30,2023
Date of decision:July 19,2023
Date of acceptance:Αugust 3,2023
Date of web publication:September 22,2023
https://doi.org/10.4103/1673-5374.385298
How to cite this article:Sharma P,Serra-Vinardell J,Introne WJ,Malicdan MCV(2024)Role of lysosomal trafficking regulator in autophagic lysosome reformation in neurons:adisease perspective.Neural Regen Res 19(5):957-958.
Open access statement:This is an open access journal,and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License,which allows others to remix,tweak,and build upon the work non-commercially,as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- From the dust: extracellular vesicles as regulators of development and neuroregeneration
- Targeting epidermal growth factor receptor signaling to facilitate cortical injury repair?
- Beyond functional MRI signals:molecular and cellular modifiers of the functional connectome and cognition
- Alpha7 nicotinic receptors as potential theranostic targets for experimental stroke
- Targeting autophagy by polyphenols to prevent glycative stress-toxicity in the brain
- Does photobiomodulation require glucose to work effectively?