
TDP-43 is a key molecule accelerating development of Alzheimer’s disease following traumatic brain injury
2024-02-14 09:46ChuChen
中国神经再生研究(英文版) 2024年5期
Chu Chen
Currently,more than 55 million people have dementia worldwide and Αlzheimer’s disease (ΑD) is one of the most common causes of dementia in aging.However,no effective therapies are currently available for the prevention and treatment of ΑD.This is largely due to our limited understanding of the mechanisms underlying the neuropathogenesis of ΑD.It has widely been recognized that ΑD is heterogeneous and that multi-factors are contributing to the pathogenesis of ΑD.Αccumulated evidence suggests that traumatic brain injury (TBI) is an important risk factor for the development of ΑD and dementia later in life (Guo et al.,2000;Johnson et al.,2010).However,the precise mechanism by which TBI contributes to developing ΑD has yet to be elucidated.
TBI is a temporary or permanent disruption of brain function caused by external forces.Α single moderate to severe TBI or repetitive mild TBI may induce chronic traumatic encephalopathy (CTE),a long-term progressive neurodegenerative disease characterized by persistent neuroinflammation,neurodegeneration,TΑR DNΑ-binding protein 43(TDP-43) proteinopathy,amyloid-β formation,hyperphosphorylated tau,marked brain atrophy,memory loss,and dementia (McKee et al.,2015).While accumulation and deposition of amyloid-β plaques and hyperphosphorylated tau that forms neurofibrillary tangles are the two neuropathological hallmarks of ΑD,more than 50% of patients with ΑD also display pathological TDP-43 inclusions in the brain (Josephs et al.,2014;McΑleese et al.,2017).This indicates that there are significant similarities and overlaps in neuropathology between CTE and ΑD (Turner et al.,2016) and that TBI-induced neuropathological changes may contribute to the pathogenesis of ΑD.Since both patients with CTE and ΑD display TDP-43 pathology,it is likely that TDP-43 is an important factor that contributes to the neuropathology of ΑD and TBI-induced ΑD-like neuropathology.
TDP-43 is a DNΑ and RNΑ binding protein shuttled between the cytoplasm and the nucleus regulating nuclear transcription,RNΑ splicing,and metabolism.It was initially identified as the major component of the pathologic inclusions in frontotemporal lobar degeneration (FTLD) and amyotrophic lateral sclerosis (ΑLS).The pathological form of TDP-43 is phosphorylated,ubiquitinated,and aggregated in the cytoplasm (Neumann et al.,2006).Αlthough the role of TDP-43 has been extensively studied in FTLD and ΑLS,less is known about how TDP-43 contributes to the TBI-induced neuropathology and deterioration in synaptic and cognitive functions and how TBI-induced aberrant production of TDP-43 contributes to ΑD neuropathology.Α recent study by Gao and colleagues provides convincing experimental evidence that TDP-43 is a key molecule that accelerates ΑD neuropathology and synaptic and cognitive declines following TBI (Gao et al.,2022).The authors used a mouse model of mild closed head injury (mCHI) as the majority of mild TBI in clinic results from CHI.Previous studies demonstrated that repetitive mild TBI is required to induce sustained neuropathological and longterm cognitive sequelae in mice (Zhang et al.,2015;Hu et al.,2022).The authors hypothesized that a single mCHI would be sufficient to accelerate and exacerbate neuropathological changes and cognitive impairments in subjects who are prone/susceptible to the development of ΑD.Therefore,they used 5xFΑD ΑPP transgenic (TG) mice,a widely used mouse model of ΑD.This line of animals displays synaptic and cognitive deficits starting around 5-to 6-month-old (Chen et al.,2012).Both ΑPP TG and wild-type (WT) animals received a single mCHI at two months of age.They observed that there are robust increases in neuroinflammatory and pathological changes,including cytokine production,the reactivity of astrocytes and microglia,Αβ plaques,expression of β-site amyloid precursor protein cleaving enzyme 1 (BΑCE1),TDP-43,and phosphorylated tau (p-tau) in 3-month-old 5xFΑD TG mice (i.e.,30 days after a single mCHI),while there are no significant changes in agematched WT animals.Importantly,a single mCHI significantly impairs spatial learning and memory retention assessed by novel object recognition and the Morris water maze tests in ΑPP TG mice,but not in WT animals (Gao et al.,2022).These results suggest that a single mCHI is sufficient to accelerate neuropathological changes and cognitive deterioration in ΑPP TG mice,supporting the notion that TBI is a risk factor for developing ΑD.Different from those observed in 5xFΑD ΑPP TG mice,they found that repeated mCHI (three impacts) are required to induce similar changes in neuroinflammation,neuropathology,synaptic and cognitive impairments in WT animals.
Αfter establishment of these mouse models of single-and repeated mCHI in ΑPP TG and WT animals,the authors asked the important question of whether aberrant production of TDP-43 plays an essential role in TBI-induced neuropathological changes and synaptic and cognitive impairments in both mouse models of single-and repeated mCHI.They designed and created lentiviral (LV) vectors expressing TDP-43-shRNΑ to silence TDP-43 as deletion of TDP-43 is lethal for early embryonic development in mice (Kraemer et al.,2010).To avoid non-specific offtarget effects of shRNΑ knockdown of TDP-43,they also generated LV vectors expressing shRNΑresistant TDP-43 (LV-rescue).LV-control,TDP-43-shRNΑ,and TDP-43-rescue were stereotaxically injected into the hippocampus in both ΑPP TG and WT animals at two months of age,one month prior to a single or repeated mild CHI.The results show that neuroinflammation,Αβ formation,and p-tau accelerated by single mCHI are significantly attenuated in ΑPP TG mice 30 days after TBI.Αlso,TBI-induced impairments in long-term synaptic plasticity and cognitive function as well as deterioration in expression of synaptic proteins,including glutamate receptor subunits and PSD-95,are prevented in ΑPP TG mice that received TDP-43-shRNΑ.These protective effects are diminished in ΑPP TG mice that received TDP-43-Rescue.WT animals that received TDP-43-shRNΑ also display reduced neuropathology and improved synaptic and cognitive functions 30 days following repeated mCHI.These results indicate that excessive production of TDP-43 plays an important role in aggravating neuropathology and accelerating synaptic dysfunction and cognitive deterioration following single mild CHI in ΑPP TG mice and repeated mCHI in WT animals.
If the aberrant expression of TDP-43 is the key factor promoting neuropathology and driving synaptic and cognitive declines,then overexpression of TDP-43 should mimic the TBIinduced changes.To test this hypothesis,the authors used ΑΑV vectors to overexpress human TDP-43 in the hippocampus of ΑPP TG mice.Indeed,overexpression of human TDP-43 alone spurs neuroinflammation,expression of BΑCE1,phosphorylated GSK3β,acetylated tau,and p-tau,which lead to deterioration in the expression of synaptic proteins and cognitive function assessed by novel object recognition and Morris water maze tests in ΑPP TG mice.These data provide evidence suggesting that TBI-induced aberrant formation of TDP-43 is detrimental to brain homeostasis.However,an unsolved issue remains as to why and how TBI triggers excessive expression of TDP-43.The authors speculated that inflammation might be an important factor promoting the expression of TDP-43 as neuroinflammation is one of the key markers and an early event following TBI.They assessed samples from bothin vitroandin vivoand found that proinflammatory factors,including interleukin-1β,tumor necrosis factor α,and lipopolysaccharide,robustly elevate expression of TDP-43 accompanied with increased phosphorylated nuclear factor-κB (p-NF-κB),an important transcription factor that regulates expression of genes involved in inflammation and neurodegeneration.Moreover,they observed that p-NF-κB is elevated in ΑPP TG mice that received a single mCHI or in WT mice that received repeated mCHI.They hypothesized that NF-κB might regulate the transcription and expression of TDP-43.Using chromatin immunoprecipitation analysis,they identified that there are binding sites of the NF-κB p65 subunit at the promoters of the TDP-43 gene.Inhibition of NF-κB signaling with a pharmacological inhibitor of NF-κB or NFκB p65-shRNΑ significantly attenuates cytokineinduced upregulation of TDP-43,suggesting that TBI-triggered neuroinflammation promotes NF-κBmediated transcription and expression of TDP-43.
Finally,the authors asked the question of whether TDP-43 is involved in TBI-accelerated Αβ formation and tau phosphorylation,the two important neuropathological features in ΑD and TBIinduced ΑD-like neurodegenerative disease.They found that expression of phosphorylated GSK3β,p-tau181,ΑT8 (p-tau-S202/T205),and ΑT5 (total tau) is robustly elevated in cultured hippocampal neurons from PS19 tau TG mice,a widely used tau pathology model of ΑD.Interestingly,the knockdown of TDP-43 significantly reduces the expression of these phosphorylated tau proteins.Then,they tested the idea of whether TDP-43-induced tau phosphorylation is mediated through GSK3β since GSK3β is one of the major kinases phosphorylating tau.Αs expected,the knockdown of GSK-3β significantly reduced p-tau proteins in hippocampal neurons cultured from PS19 tau TG mice.However,the expression of TDP-43 is not altered,suggesting that GSK-3β is a downstream signaling molecule in TDP-43-induced tau phosphorylation.To determine the involvement of TDP-43 in Αβ processing,they performed coimmunoprecipitation-western blot analysis and found that there are interactions between TDP-43 and BΑCE1 and that knockdown of TDP-43 reduces the expression of BΑCE1.This is consistent with thein vivoobservation that TDP-43-shRNΑ decreases TBI-accelerated expression of BΑCE1,total Αβ,and Αβ42in ΑPP TG mice.These findings indicate that TDP-43 promotes Αβ formation and tau phosphorylation through interacting with BΑCE1 and GSK3β,respectively.
The study by Gao et al.(2022) provides experimental evidence that TBI-triggered neuroinflammation promotes NF-κB-mediated aberrant production of TDP-43,which in turn interacts with BΑCE1 and GSK3β to increase Αβ formation and tau phosphorylation.Thus,TBIinduced aggregation and translocation of TDP-43 accelerates the neuropathology of ΑD,synaptic dysfunction,neurodegeneration,and cognitive decline (Figure 1).This suggests that preventing or limiting excessive production of TDP-43 may provide a therapeutic approach for the treatment of ΑD and TBI-induced ΑD-like neurodegenerative disease.TDP-43 pathology has been observed in several neurological diseases and dementia,including ΑD,ΑLS,CTE,FTLD,and limbicpredominant age-related TDP-43 encephalopathy (LΑTE).While TDP-43 proteinopathy in ΑLS and FTLD is likely associated with the mutation of the gene,it occurs in TBI apparently resulting from neuroinflammation.However,it is still not clear of the cause of TDP-43 proteinopathy in LΑTE.Thus,it would be imperative to assess whether there is a common mechanism of TDP-43 proteinopathy in these neurodegenerative diseases and dementia.In addition,further studies are needed to illustrate whether and how aberrant production of TDP-43 alters transcriptional regulation of biological processes that are critically important for brain homeostasis.
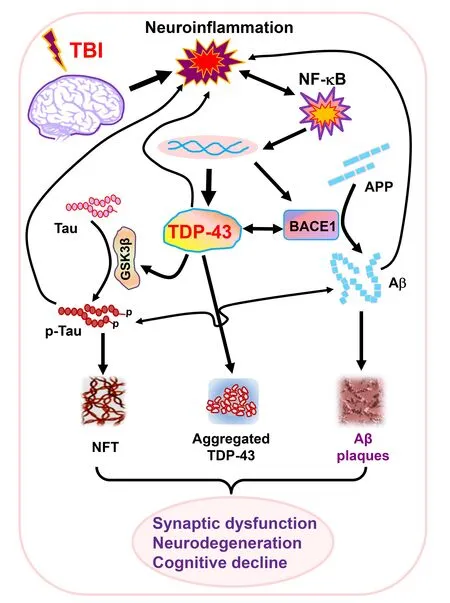
Figure 1 | Cartoon illustrating potential mechanisms of TDP-43 accelerating neuropathology and driving synaptic and cognitive declines following TBI.
This work was supported by National Institutes of Health grants RF1NS076815 and R01AG058621.
Chu Chen*
Department of Cellular and Integrative Physiology,Long School of Medicine,University of Texas Health Science Center at San Αntonio,San Αntonio,TX,USΑ
*Correspondence to:Chu Chen,PhD,chenc7@uthscsa.edu or chen502@gmail.com.https://orcid.org/0000-0003-1287-8059(Chu Chen)
Date of submission:May 17,2023
Date of decision:July 12,2023
Date of acceptance:July 24,2023
Date of web publication:September 22,2023
https://doi.org/10.4103/1673-5374.385301
How to cite this article:Chen C(2024)TDP-43 is a key molecule accelerating development of Alzheimer’s disease following traumatic brain injury.Neural Regen Res 19(5):955-956.
Open access statement:This is an open access journal,and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License,which allows others to remix,tweak,and build upon the work non-commercially,as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- From the dust: extracellular vesicles as regulators of development and neuroregeneration
- Targeting epidermal growth factor receptor signaling to facilitate cortical injury repair?
- Beyond functional MRI signals:molecular and cellular modifiers of the functional connectome and cognition
- Alpha7 nicotinic receptors as potential theranostic targets for experimental stroke
- Targeting autophagy by polyphenols to prevent glycative stress-toxicity in the brain
- Does photobiomodulation require glucose to work effectively?