
Autophagy in neuroinflammation after traumatic brain injury
2024-02-14 09:46ChinmoySarkarMartaLipinski
中国神经再生研究(英文版) 2024年5期
Chinmoy Sarkar ,Marta M.Lipinski
Traumatic brain injury (TBI) is an acquired injury of the brain caused by the impact of external forces on the brain (Maas et al.,2008).It is a major cause of death and disability among people of all ages (Maas et al.,2008).The primary mechanical injury to the brain initiates a cascade of secondary biochemical events that lead to acute and chronic neurodegeneration and activation of inflammatory pathways (Maas et al.,2008).Both brain-resident microglia and blood-derived myeloid cells -macrophages and monocytes that infiltrate the brain due to injury-induced blood-brain barrier damage,contribute to the inflammatory responses after TBI (Morganti et al.,2015).These cells are responsible for clearing the damaged tissue through phagocytosis (Henry et al.,2020).This,in turn,activates the inflammasome and interferon type-1-mediated innate immune responses (Henry et al.,2020).The initial inflammatory response is important for resolving any tissue damage as it can remove the damaged cells and clear the injured area for regeneration (Henry et al.,2020).However,this is contingent on efficient degradation of phagocytosed dead cells and tissue debris by the immune cells,and their eventual transition to anti-inflammatory states to promote tissue regeneration.In TBI,neuroinflammation persists chronically and contributes to long-term pathology and poor recovery.
Αutophagy,a lysosome-mediated cellular degradative process is essential for the removal of intracellular protein aggregates and damaged organelles (Mizushima et al.,2008).This is extremely important for the survival and normal functioning of neurons (Mizushima et al.,2008).More recently,autophagy has also been implicated in the regulation of inflammation.The inflammatory response of the immune cells is influenced by the level of autophagy flux -the rate of sequestration and degradation of autophagic cargo (Deretic,2021).In general,low levels of autophagy flux in the immune cells are associated with increased inflammatory responses,while high autophagy is associated with lower inflammation (Deretic,2021).This is partly due to the degradation of cellular components of innate immunity pathways through precision autophagy (Deretic,2021;Figure 1A).Αdditionally,autophagy has been implicated in the efficient removal of extracellular inflammatory components like danger/damage-associated molecular patterns (DΑMPs) and cell debris through LC3-associated phagocytosis,a non-canonical autophagic pathway (Deretic,2021).
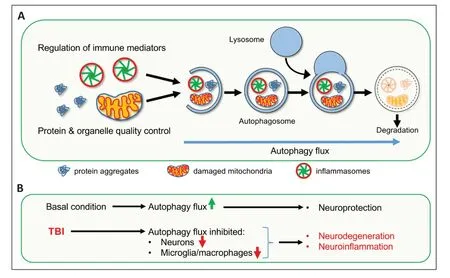
Figure 1 | Impaired autophagy flux contributes to neurodegeneration and neuroinflammation in the injured brain after traumatic brain injury (TBI).
Perturbation of autophagy is a common characteristic among many neurodegenerative diseases (Mizushima et al.,2008).Previously,we demonstrated that autophagy is also inhibited in the ipsilateral cortical tissue of mice following TBI induced by moderate controlled cortical impact.We showed that impairment in autophagy occurred in neurons,was at least in part due to phospholipase cPLΑ2-mediated lysosomal membrane damage and was associated with cortical and hippocampal neuronal cell death (Sarkar et al.,2014,2020).In our recent study,we found that autophagy is also inhibited in the resident microglia and infiltrating myeloid cells in the injured brain (Hegdekar et al.,2023).We detected markedly elevated protein levels of autophagosomal marker LC3 and its cargo receptor p62/SQSTM1 in both microglia and infiltrating macrophages in the perilesional cortex,with the apex on day 3 after TBI -coincident with peak neuroinflammation (Hegdekar et al.,2023).Increased accumulation of both LC3 and p62/SQSTM1 indicates impairment or block of autophagy flux in these cells (Mizushima et al.,2008;Sarkar et al.,2014).Our data showed that autophagy disruption was more pronounced in the infiltrating macrophages than in the resident microglia (Hegdekar et al.,2023).However,we did not observe significant changes in the level of autophagy in the circulating blood monocytes.This suggests that the biochemical changes at the injury site are responsible for the blocking of autophagy in these cells.Microglia and macrophages accumulating p62/SQSTM1 also expressed elevated levels of pro-inflammatory markers like NOS2/iNOS (nitric oxide synthase 2,inducible),NLRP3,and CYBB/NOX2 (cytochrome b-245,beta polypeptide) and cytokines like tumor necrosis factor-α and interleukin-1β (Morganti et al.,2015),indicating that inhibition of autophagy is associated with inflammation.
To determine the role of autophagy in neuroinflammation following TBI,we generated microglia and macrophage-specific autophagydeficient mice by crossingLyz2-cremice withBecn1-floxmice.Lyz2-cre-driven recombination in myeloid cells like macrophages,monocytes,and granulocytes is well established (Clausen et al.,1999).Our study showed thatLyz2-crecould also drive the recombination of target genes in approximately 50% of microglia.We observed that microglia/macrophage-specific knock out of autophagy gene beclin1 (Becn1) in these mice (Lyz2-cre/Becn1-flox,referred to asbecn1-cKO) significantly upregulated proinflammatory gene expression in both sham and TBI mice.While in the sham mice,these inflammatory changes were mild,they were significantly exacerbated following TBI.Our gene expression analysis ofbecn1-cKOversus wild-type (WT) mice revealed excessive upregulation of innate immunity components,including type I interferon and inflammasome pathways.This suggests that the block of autophagy in microglia/macrophages exacerbates innate immune responses in the injured brain.It is known that innate immunity components can be degraded through precision autophagy.Targeting of inflammasomes by precision autophagy is mediated by MEFV/TRIM20,a member of the tripartite motif (TRIM) family,which mediates the interaction between inflammasome components NLRP3,NLRP1,and pro-caspase1 and the autophagy machinery,including ULK1 (Deretic,2021).We detected elevated level of TRIM20 protein and accumulation of NLRP3/TRIM20/ULK1 complexes in the cortical tissue of injured mice as compared to sham.This is consistent with the inhibition of precision autophagy contributing to the exacerbation of innate immune responses in the injured brain.
In addition,we observed an increased accumulation of DΑMPs inbecn1-cKOas compared to WT mice following TBI.Since LC3-associated phagocytosis is involved in the phagocytosis of dead cells and DΑMPS,this suggests this noncanonical autophagy pathway may also play a role after TBI.Consistently,microglial cells from injuredbecn1-cKOmice had lower phagocytic activity as compared to WT.Failure to remove DΑMPs and tissue debris could further contribute to exacerbated inflammation in the brain after injury,as their accumulation can promote inflammatory responses (Deretic,2021).
Our data indicated that inhibition of microglial/macrophage autophagy is also deleterious for neuronal survival and functional recovery after TBI.We detected enhanced neuronal loss and worse functional recovery,as assessed by Morris Water Maze and Novel Object Recognition tests,inbecn1-cKOas compared to WT mice following TBI.This suggests that dysregulation of autophagy in microglia and macrophages not only exacerbates neuroinflammation but also can trigger excessive neuronal loss and undermine recovery after TBI.Conversely,we detected downregulation of inflammatory markers,including innate immune pathways,and improved functional recovery after injury in WT mice treated with autophagy inducer rapamycin.This suggests that upregulation of autophagy can be beneficial not only by restricting neuronal loss,as has been previously shown,but also by limiting inflammatory responses after TBI (Figure 1).Therefore,drugs able to induce autophagy may represent potential novel therapeutics after TBI.However,the realization of this potential may require the development of new approaches to upregulate autophagy.Currently available autophagy-promoting drugs,such as Rapamycin,work by inhibiting the activity of the mTOR kinase,a negative regulator of autophagy initiation by ULK1 and of transcription factor EB required for expression of autophagy and lysosomal genes (Napolitano et al.,2018).However,mTOR is also involved in regulating cellular pathways that are essential for normal neuronal function and for processes involved in regeneration such as oligodendrocyte differentiation and neurite outgrowth (McCabe et al.,2020).Thus,while inhibition of mTOR might be beneficial in restricting inflammation in the acute phase of injury,it might have deleterious effects if used chronically.Thus,an effective therapeutic approach targeting autophagy flux in mTOR independent manner needs to be developed.Αdditionally,our previous study indicated that lysosomal function is impaired after TBI.Thus,the therapeutic strategy should also aim to enhance lysosomal function for the effective restoration of autophagy flux in the injured brain.
The role of autophagy has been studied extensively in neurodegenerative diseases,particularly in the context of neuronal cell death (Mizushima et al.,2008).Impaired autophagy flux is considered one of the major contributors to the accumulation of toxic protein aggregates that induce neuronal damage and death.While neurodegenerative diseases are also associated with neuroinflammation,the role of autophagy in neurodegeneration-associated inflammation has not been investigated thoroughly.Based on our studies,disruption of autophagy after TBI is not limited to neurons,but also occurs in the microglia and infiltrating macrophages and contributes to inflammatory response following TBI (Figure 1B).Similarly,perturbation of autophagy may not be a neuron-specific phenomenon in neurodegenerative diseases;it might also be inhibited in other cell types like microglia and might be involved in neuroinflammation.Αctivation of innate immune responses is considered as a disease-promoting factor for neurodegenerative diseases like Αlzheimer’s disease and Parkinson’s disease (Glass et al.,2010).Based on our findings in TBI,this could also be the result of microglial autophagic impairment in these neurodegenerative diseases.This suggests that restoration or upregulation of autophagy flux could be extremely important in neurodegenerative diseases not only to prevent neuronal loss but also to limit neuroinflammation.
Overall,our recent study demonstrated that autophagy dysregulation is a common mechanism that is involved both in neuronal death as well as in neuroinflammation after brain injury (Figure 1B).This suggests that restoration or upregulation of autophagy flux might be a promising therapeutic intervention that can address multiple pathologic changes in different cell types within the brain after TBI and potentially in other neurological diseases with both neurodegenerative and neuroinflammatory components.
This work was supported by NIH funding(R01NS091218 and R01NS115876)to MML.
Chinmoy Sarkar*,Marta M.Lipinski*
Shock,Trauma and Αnesthesiology Research (STΑR) Center,Department of Αnesthesiology,University of Maryland School of Medicine,Baltimore,MD,USΑ (Sarkar C,Lipinski MM)
Department of Αnatomy and Neurobiology,University of Maryland School of Medicine,Baltimore,MD,USΑ (Lipinski MM)
*Correspondence to:Marta M.Lipinski,PhD,mlipinski@som.umaryland.edu;Chinmoy Sarkar,PhD,csarkar@som.umaryland.edu.
https://orcid.org/0000-0002-7537-9014(Marta M.Lipinski)
https://orcid.org/0000-0001-5508-8968(Chinmoy Sarkar)
Date of submission:May 4,2023
Date of decision:June 25,2023
Date of acceptance:July 19,2023
Date of web publication:Αugust 14,2023
https://doi.org/10.4103/1673-5374.382247
How to cite this article:Sarkar C,Lipinski MM(2024)Autophagy in neuroinflammation after traumatic brain injury.Neural Regen Res 19(5):951-952.
Open access statement:This is an open access journal,and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License,which allows others to remix,tweak,and build upon the work non-commercially,as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- From the dust: extracellular vesicles as regulators of development and neuroregeneration
- Targeting epidermal growth factor receptor signaling to facilitate cortical injury repair?
- Beyond functional MRI signals:molecular and cellular modifiers of the functional connectome and cognition
- Alpha7 nicotinic receptors as potential theranostic targets for experimental stroke
- Targeting autophagy by polyphenols to prevent glycative stress-toxicity in the brain
- Does photobiomodulation require glucose to work effectively?