
超高效液相色谱-串联质谱法同时测定5种抗逆转录病毒药物血药浓度*
2024-01-31 13:18张晓颖叶珍洁吴灵洁原津津俞晓玲
医药导报 2024年2期
张晓颖,叶珍洁,吴灵洁,原津津,俞晓玲,4
(福建医科大学孟超肝胆医院 1.精准药学实验室;2.重点实验室;3.感染科;4.药学部,福州 350025)
据世界卫生组织(World Health Organization,WHO)估计,截至2022年底,全球现存活获得性人类免疫缺陷病毒(human immunodeficiency virus,HIV)感染/艾滋病患者3 900万例,艾滋病相关死亡的人数达4 040万例,有2 980万例正在接受抗病毒治疗(antiretroviral therapy,ART),ART全球覆盖率达到76%[1],ART是国际上公认的防治艾滋病最有效的方法。目前,应用于临床的抗逆转录病毒药物分为6大类,①核苷类逆转录酶抑制剂:以拉米夫定(lamivudine,3TC)、替诺福韦(tenofovir,TDF)为代表;②非核苷类逆转录酶抑制剂:如奈韦拉平、依非韦伦(efavirenz,EFV)等;③蛋白酶抑制剂:如克力芝、利托那韦等;④融合抑制剂:艾博韦泰;⑤CC趋化因子受体5拮抗剂;⑥整合酶抑制剂:以多替拉韦(dolutegravir,DTG)、拉替拉韦(raltegravir,RAL)为代表[2]。其中多替拉韦、拉替拉韦、依非韦伦、拉米夫定和替诺福韦是治疗HIV的一线药物,国内外艾滋病诊疗指南均推荐拉米夫定+替诺福韦+多替拉韦/拉替拉韦/依非韦伦作为成人艾滋病初治患者的ART方案之一[3-4]。然而,大部分抗HIV药物的血药浓度具有显著的个体间差异,往往因为疗效不足或无法忍受的不良反应调整剂量甚至停药。基因多态性、肝/肾功能不全、年龄、食物和药物相互作用均会导致抗病毒药血药浓度范围变异性大[5]。多替拉韦、拉替拉韦、依非韦伦、拉米夫定和替诺福韦的血浆谷浓度与疗效、不良反应显著相关,谷浓度过高易发生不良反应,浓度不足则易出现病毒学失败或病毒耐药变异[6]。故指南[3]和文献[6]均明确指出需对抗HIV病毒药物开展治疗药物监测(therapeutic drug monitoring,TDM),指导个体用药剂量,从而达到增效减毒的目的。
在抗病毒药的药动学或TDM研究中,超高效液相色谱-串联质谱(ultra high performance liquid chromatography-tandem mass spectrometry,UPLC-MS/MS)法因其高效、高选择性、高灵敏度、适用范围广等优点,越来越广泛应用于体内生物样品分析领域。此外,MS/MS能够实现多个成分同时定性、定量分析,降低样品用量与分析检测时间,大大提升检测效率,现已成为血药浓度检测的“金标准”。目前,国内外仅有一项研究采用LC-MS/MS法同时测定本研究的5种抗HIV病毒药物及其他抗病毒药[7],但该方法分析时间较长(>6 min),样品制备过程耗时长(>10 min)。鉴于此,本研究旨在建立UPLC-MS/MS法同时测定多替拉韦、拉替拉韦、依非韦伦、替诺福韦和拉米夫定血药浓度,并应用于临床样本检测,为TDM提供技术依据。
1 材料与试药
1.1仪器 LC-30A超高效液相色谱仪(日本Shimadzu公司);AB SCIEX TRIPLE QUAD TM 5500三重四级串联质谱仪(美国AB SCIEX公司);IKA VIBRAX VXR BASIC圆周振荡器(德国IKA公司);Eppendorf centrifuge 5810R高速冷冻离心机(德国Eppendorf公司);KQ3200E超声波清洗器(昆山市超声仪器厂);MSE225S-CE电子分析天平(德国Sartorius公司,感量:0.01 mg)。
1.2药品与试剂 多替拉韦对照品(含量:98%,批号:1-TIM-1-1)、拉替拉韦对照品(含量:96%,批号:27-MJC-148-1)、依非韦伦对照品(含量:97%,批号:7-ARD-114-5)、拉米夫定(含量:98%,批号:2-SEH-171-1)、替诺福韦(含量:96%,批号:1-JMO-90-1)均购自Toronto Reseach Chemicals Inc;同位素内标:多替拉韦-D5(含量:97.5%,批号:13576)、拉替拉韦-D4(含量:96.9%,批号:13765)、依非韦伦-D5(含量:94.6%,批号:13656)、拉米夫定-13C-15N2(含量:96.2%,批号:13575)、替诺福韦-D7(含量:99.8%,批号:13574)均购自于Quality Control Solutions Inc;甲酸(AR级,西陇科学股份有限公司);甲醇、乙腈(HPLC级,德国Merck KGaA);去离子水由我院血透室超纯水净化仪制。
2 方法与结果
2.1LC-MS/MS分析条件
2.1.1色谱条件 色谱柱Shim-pack XR-ODS Ⅲ(2.0 mm×50 mm,1.6 μm),预柱Shim-pack GIST-HP(G)C18(2.1 mm×10 mm,2 μm) ;流动相:A为0.1%甲酸,B为0.1%甲酸乙腈,梯度洗脱程序如下(0—0.5 min,5%B;0.5—1.0 min,5% → 90%B;1.0—4.0 min,90%B;4.0—4.01 min,90%→5%B;4.01—5.0 min,5%B);流速为0.3 mL·min-1,进样量为2 μL,自动进样器温度为4 ℃,柱温箱温度为40 ℃。
2.1.2质谱条件 采用电喷雾离子源(electric spray ion source,ESI),正离子模式扫描,离子源电压为5 500 V,离子源温度为550 ℃,气帘气压力、碰撞气压力、辅助气1压力和辅助气2压力分别设置为40、9、50、50。扫描方式为多反应监测模式(multiple reaction monitoring mode,MRM),5个分析物及内标的质谱参数见表1。
2.2对照品储备液及工作液的配制 精密称取2份多替拉韦、拉替拉韦、依非韦伦、拉米夫定、替诺福韦对照品适量,均加入50%甲醇溶解,转移至10 mL棕色量瓶中,定容至刻度,并储存于-30 ℃冰箱保存备用。多替拉韦的校准曲线和QC储备液浓度分别为179.7、214 μg·mL-1、拉替拉韦储备液浓度分别为375.6、151 μg·mL-1、依非韦伦储备液浓度分别为252.6、114.7 μg·mL-1、拉米夫定储备液浓度分别为256、343.7 μg·mL-1、替诺福韦储备液浓度分别为125、117.3 μg·mL-1。
精密吸取各分析物的标准曲线储备液适量,加入50%甲醇稀释混合,制成系列浓度的标准曲线工作液(多替拉韦:0.625、1.25、2.5、5、10、15、20、30 μg·mL-1;拉替拉韦、拉米夫定、替诺福韦:0.1、0.2、0.4、0.8、1.6、2.5、3.2、5 μg·mL-1;依非韦伦:1.25、2.5、5、10、20、30、40、60 μg·mL-1)。同时,精密吸取各分析物的QC储备液适量,按照上述方法配制定量下限(lower limit of quantitation,LLOQ)、低(QL)、中(QM)、高(QH)四水平质控样品工作液,多替拉韦质控质量浓度分别为0.625、1.5、6、24 μg·mL-1;拉替拉韦、拉米夫定、替诺福韦:0.1、0.25、1、4 μg·mL-1;依非韦伦:1.25、3、12、48 μg·mL-1。上述工作液均储存于-30 ℃冰箱保存备用。
2.3内标储备液及工作液的配制 取内标多替拉韦-D5、拉替拉韦-D4、依非韦伦-D5、拉米夫定-13C-15N2、替诺福韦-D7适量溶于10 mL棕色量瓶中,加50%甲醇溶解并定容,配制成质量浓度为均为100 μg·mL-1的内标储备液。取上述5种内标储备液适量,用50%甲醇继续稀释,配制成含多替拉韦-D5、拉替拉韦-D4、依非韦伦-D5、拉米夫定-13C-15N2、替诺福韦-D7的质量浓度分别为10、0.8、30、1.6、2.5 μg·mL-1的混合内标工作液。
2.4血浆样品预处理 取待测血浆样品100 μL(标准曲线工作液/质控工作液10 μL+空白血浆90 μL),加入内标工作液10 μL,先后加入纯水100 μL和乙腈500 μL,涡旋振荡1 min,4 ℃、12 000×g离心5 min后,取上清液300 μL,加入乙腈-水(50:50)300 μL稀释,振动30 s,混匀后过孔径0.45μm微孔滤膜,取2 μL进样分析。
2.5方法学考察
2.5.1专属性实验 取6份不同来源的空白血浆,以等体积50%甲醇替代内标工作液,按“2.4节”下方法操作,得到空白血浆色谱图;另取6份空白血浆制成含分析物的标准血浆溶液,按“2.4节”下方法操作,获得含分析物与内标样品的色谱图。空白血浆中干扰组分的响应低于分析物定量下限响应的20%,并低于内标响应的5%[8-10]。结果见图1所示,5个分析物及同位素内标的峰型良好,且空白血浆中干扰组分的响应远低于分析物定量下限响应的20%、内标响应的5%,证实了所开发的方法对分析物和同位素内标具有选择性和特异性。

图1 空白血浆(A)、LLOQ样品中分析物(B)和内标的色谱图(C)
2.5.2标准曲线及LLOQ 取空白血浆90 μL置于1.5 mL EP 管中,加入系列含分析物的标准曲线工作液10 μL,配制成系列标准曲线样品和质控样品,按“2.4节”下方法操作。以分析物与内标峰面积比值(Y)为纵坐标,分析物浓度(X,ng·mL-1)为横坐标,采用加权1/x2最小二乘法进行线性回归分析,获得各分析物的线性回归方程。结果表明,DTG、EFV、RAL、3TC和TDF血药浓度分别在62.5~3 000、125~6 000、10~500、10~500、10~500 ng·mL-1范围内线性关系良好,相关系数(R2)在0.998~0.999。5个分析物的各浓度水平准确性均为89.8%~109.9%范围内,各分析物LLOQ的S/N均>5,符合“生物样品定量分析方法验证指导原则”的要求[8],色谱图见图1。
2.5.3准确度与精密度 取空白血浆90 μL,加入质控工作液10 μL,配制 LLOQ、QL、QM、QH 4个浓度的质控样品,每种浓度平行制备6份,按“2.4节”下方法操作,计算日内准确度和精密度,连续测定3 d,确定日间准确度和精密度(RSD)。各质控样本(除LLOQ外)日内和日间的准确度、RSD一般应在标示值的±15%内,LLOQ的准确度、RSD应在标示值的±20%范围内[8-10]。DTG、RAL、EFV、3TC和TDF的日内、日间准确度和精密度结果见表2,5个分析物各浓度水平的日内及日间精密度RSD值均<7%,日内和日间各浓度水平的准确度为94.0%~109.3%,符合“生物样品定量分析方法验证指导原则”的要求[8]。

表2 5个分析物在血浆中的日内、日间精密度和准确度
2.5.4提取回收率和基质效应试验 按照“2.4节”下方法分别制备高、中、低浓度QC样品进样分析,以对照品和内标峰面积比值作为A1;取6批不同来源的空白血浆,按照“2.4节”方法进行血浆样品前处理,取全部上清液分别加入高、低浓度质控工作液和内标工作液各10 μL,再按“2.4节”下方法稀释进样分析,以对照品和内标峰面积的比值作为A2;以纯水代替空白血浆,按“2.4节”操作配制成高、低QC水平进样分析,以对照品和内标峰面积的比值作为A3。A1/A2 为提取回收率(extraction recovery,ER),A2/A3 为基质效应(matrix effect,ME)。同时,采用同样的方法分析溶血及高脂血浆的基质效应。分别计算5个分析物及其同位素内标的基质效应,进而计算内标校正的基质效应,6批基质内标校正的基质效应变异系数≤15%[8-10]。结果见表3,5个分析物及其内标的提取回收率为98.7%~104.5%,RSD均<5%;6批不同来源正常血浆的基质效应为98.1%~106.0%,RSD均<6%;溶血及高脂血浆的基质效应为95.7%~105.8%,RSD均<5%。因此,空白、溶血及高脂血浆基质效应不影响各分析物的定量。

表3 5个分析物在血浆中的基质效应和提取回收率
2.5.5残留试验 在进样高浓度样品(ULOQ)后,通过分析空白血浆样本来估计残留。根据《中华人民共和国药典》2020年版规定,分析高浓度样品之后,空白样品中的残留应不超过LLOQ的20%,且不超过内标的5%[8-10]。研究结果显示,LLOQ中DTG、LMV、TNF的残留<10%,RAL、EFV并无残留;所有同位素内标的残留均<1.3%,故各分析物的残留不影响检测的准确度和精密度。
2.5.6稀释效应 配制高于定量上限浓度的血浆样本(多替拉韦、拉替拉韦、依非韦伦、拉米夫定及替诺福韦质量浓度分别为80、8、120、8、8 μg·mL-1),并用空白血浆稀释该样品,稀释倍数分别为4、10、40倍(每个稀释因子平行制备6份)。样品稀释不应影响准确度和精密度,稀释后样本的准确度和精密度应在±15%之内[8-10]。结果显示,稀释4、10、40倍后,样本的准确度为93.8%~113.3%,精密度为2.4%~5.7%。因此,浓度超过定量上限的血浆样本可以在样本前处理和分析前稀释。
2.5.7稳定性试验 平行配制QC样品(QL、QH)各6份,考察多替拉韦、拉替拉韦、依非韦伦、拉米夫定和替诺福韦在不同储存或工艺条件下的血浆样品稳定性。
所有的稳定性结果均是通过与0时刻比较而获得,每个水平的QC浓度均值与标示浓度的偏差应在±15%范围内[8-10]。各分析物的稳定性结果见表4,5个分析物的血浆样品在室温下可稳定放置12 h、4 ℃可放置2 d、-30 ℃可放置20 d、可反复冻融3次、自动进样室(4 ℃)中可持续放置12 h、重复进样3次依旧保持稳定,各质控样品在上述条件下的RSD值均<10%。

表4 5个分析物在血浆中的稳定性
2.6临床应用 该研究已通过福建医科大学孟超肝胆医院伦理委员会批准(伦理号:科审2023_005_01)。通过监测服用多替拉韦、拉替拉韦、依非韦伦、拉米夫定或替诺福韦的HIV患者稳态血浆谷浓度,检验本方法的适用性。多替拉韦、拉替拉韦、依非韦伦、拉米夫定和替诺福韦的消除半衰期分别为14、9、52~76、5~7、17 h,以上抗病毒药在4~5个半衰期后达到稳态血药浓度。考虑到个体间的药动学差异,测定服用多替拉韦、拉替拉韦、拉米夫定或替诺福韦至少1周后的稳态血浆谷浓度及依非韦伦至少16 d后的稳态血浆谷浓度。患者血药浓度达稳态后,在下一次给药前30 min抽取静脉血2 mL于EDTA2K采血管中,3 000×g离心5 min,将上清液转移至1.5 mL EP管中,于-30 ℃下保存至分析检测。本研究共检测样本29例,其血药浓度结果见表5。本方法的定量浓度范围可基本覆盖多替拉韦、拉米夫定、替诺福韦的临床样本检测需求。后续会继续收集更多的临床样本进行检测,为HIV患者制定个体化治疗提供参考。
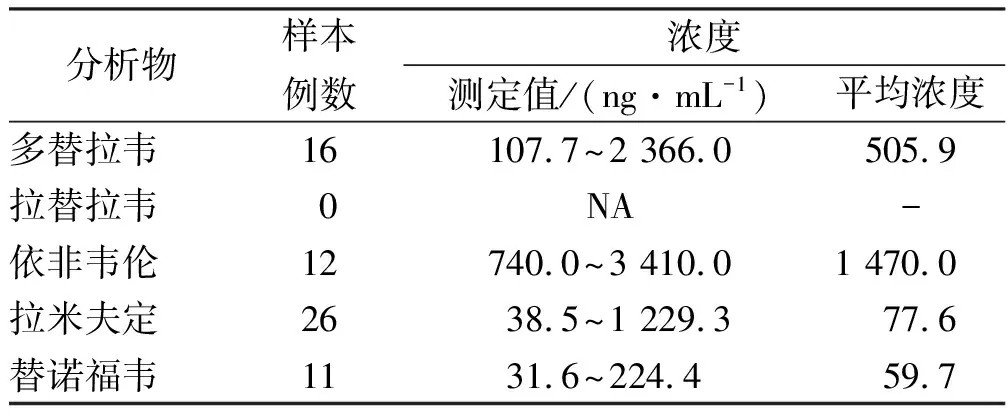
表5 HIV患者稳态血药谷浓度
3 讨论
国内外已有文献报道关于5个分析物单独或与其他药物同时测定检测方法,如高效液相色谱-紫外检测法(HPLC-UV)、毛细管电泳法和UPLC-MS/MS等[11]。但毛细管电泳-UV法[12-13]和HPLC-UV法[14-15]检测灵敏度均较低,易受基质中杂质干扰、分析时间较长(>20 min),样品前处理复杂,二者均不适用于HIV患者多种抗病毒药物的高通量分析。UPLC-MS/MS结合色谱的高分离效率、质谱的高选择性、高灵敏度等优点,成为临床上血药浓度检测的金标准。SINGH等[16]采用LC-MS/MS法测定人血浆中阿巴卡韦/多替拉韦/拉米夫定的血药浓度,但该方法使用缓冲盐作为流动相,配制繁琐、费时且易加速消耗色谱柱的使用寿命;PENCHALA等[17]采用液液萃取法、固相萃取法进行前处理,但操作复杂,耗时较长且浪费有机溶剂。仅国外一项研究同时检测5种分析物及其他抗病毒药[7],但该方法分析时间较长(>6 min),样品制备过程耗时长(>10 min),且国内尚无5种分析物同时测定的报告。基于此,结合已有的文献方法,本研究从色谱、质谱条件及血样前处理方法进行优化。
本研究考察不同流动相及梯度洗脱程序,将纯水、0.1%甲酸、2 mmol·L-1乙酸铵水溶液作为水相分别与甲醇、乙腈、0.1%甲酸甲醇、0.1%甲酸乙腈等有机相进行配比,结果显示甲醇中替诺福韦峰形拖尾,拉米夫定、替诺福韦在不加酸的流动相中不出峰,而拉替拉韦、依非韦伦在加入乙酸铵的流动相中后响应变低,后基线变高,故最终采用0.1%甲酸-0.1%甲酸乙腈作为流动相,确保各分析物峰型良好、响应高及保留时间短。拉米夫定和替诺福韦的极性强,而多替拉韦、拉替拉韦和依非韦伦的极性较弱,故初始流动相水相比例升高有助于增加拉米夫定和替诺福韦保留时间,增大二者响应。在样本的前处理上,本方法选择蛋白沉淀法,对比甲醇,乙腈作为沉淀剂,5个分析物的响应更高。但5个分析物的极性差异较大,前处理单纯采用乙腈沉淀,拉米夫定和替诺福韦难以达到理想的提取效果,故在乙腈沉淀前加适量纯水,增大拉米夫定和替诺福韦的溶解度,可大大提高二者的质谱响应。同时,采用同位素内标来降低血浆中内源性物质对响应造成的直接或间接的影响。相较于其他方法,本方法运行时间短(5 min),以0.1%甲酸-0.1%甲酸乙腈作为流动相,不影响色谱柱的寿命,血浆样本前处理简单、低成本,满足临床高通量检测需求。
本研究建立的人血浆中多替拉韦、拉替拉韦、依非韦伦、拉米夫定和替诺福韦同时测定方法简便、快速、高效、准确度高、线性范围广,适用于HIV患者的多替拉韦、拉替拉韦、依非韦伦、拉米夫定和替诺福韦的治疗性药物监测研究,为HIV患者制定个体化的用药方案提供依据。
猜你喜欢
传染病信息(2022年6期)2023-01-12
临床合理用药杂志(2021年5期)2021-04-17
昆明医科大学学报(2021年1期)2021-02-07
肝博士(2020年5期)2021-01-18
中华养生保健(2020年5期)2020-11-16
制造技术与机床(2017年9期)2017-11-27
当代医药论丛(2017年22期)2017-04-12
西南石油大学学报(自然科学版)(2015年3期)2015-04-16
中国当代医药(2015年17期)2015-03-01
疑难病杂志(2014年12期)2014-04-16
