
基于药物拼接的verubulin 衍生物的设计合成及活性评价
2024-01-27 16:33王晓锋尹东锋
西北药学杂志 2024年1期
王晓锋,王 明,张 晨,李 倩,尹东锋
新疆军区总医院药剂科,乌鲁木齐 830000
微管是由α、β微管蛋白动态装配而成的长丝状管状结构,广泛存在于真核细胞中[1]。微管蛋白通过与不同的调节因子结合参与细胞形态维持、细胞增殖、物质运输及信号转导等过程,是抗肿瘤药物的重要靶点之一[2-3]。,由于作用于微管蛋白秋水仙碱结合位点的化合物结构多样,且能克服肿瘤细胞的耐药性并破坏已形成的肿瘤血管[4],故而逐渐成为研究的热点。
verubulin(化合物1,结构见图1)是最早发现含有喹唑啉结构的作用于微管蛋白秋水仙碱结合位点的微管聚集抑制剂,在体外对多种肿瘤细胞表现出很强的增殖抑制活性[5-9]。BANERJEE S 等[10]通过解析化合物1 的衍生物(2)(结构见图1)与微管蛋白复合晶体的结构(PDB 编码:6BR1),揭示了该类化合物的作用方式,为进一步进行结构修饰奠定了基础。MP-HJ-1c(化合物3,结构见图1)是新发现的另一类作用于微管蛋白秋水仙碱结合位点的化合物[11-12],与1 及秋水仙碱[13]、康普瑞汀(CA-4)[14]的结合位点相比,其结构中的吡咯并[2,3-d]噻唑-5-甲酰胺基团,“伸入”微管蛋白β亚基更深的空腔中,与βN165、βE198 和βV236 形成氢键(PDB 编码:5YZ3)。通过将6BR1 和5YZ3 进行叠合,设想在1 结构中喹唑啉环2 位引入吡咯并[2,3-d]噻唑-5-甲酰胺结构,由此设计了化合物4a 和4b(结构见图1),对其分别进行了化学合成和初步体外活性评价。

图1 化合物1~4 的结构Fig.1 Structure of compounds 1-4
1 仪器与材料
1.1 仪器
磁力搅拌器(德国IKA 公司);RY-Ⅰ型熔点仪(天津市天分分析仪器厂);AVANCE Ⅲ HD600 型超导核磁共振仪(德国BRUKER 公司);JNM-ECA-400 型超导核磁共振仪(日本电子株式会社);Xevo G2-XS QTOF 型高分辨质谱仪(沃特世公司);分子模拟软件Discovery Studio 3.0(Accelrys,San Diego,USA)。
1.2 试药
5-噻唑甲醛(质量分数为98%)、叠氮乙酸乙酯(质量分数为98%)、5 mol·L-1乙醇钠溶液(质量分数为99%)、氢氧化锂(质量分数为98%)、N-Boc-甘氨酸(质量分数为98%)、N-Boc-β-丙氨酸(质量分数为98%)、2-氨基苯甲酰胺(质量分数为98%)、N,N-二异丙基乙基胺(质量分数为99%)、1-乙基-(3-二甲基氨基丙基)碳化二亚胺盐酸盐[1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochlorid,EDCI·HCl,质量分数为98%]、1-羟基苯并三唑(1-hydroxybenzotriazole,HOBt,质量分数为99%)、三乙胺(质量分数为98%)、N,N-二甲基-4-氨基吡啶(质量分数为98%)、对甲苯磺酰氯(质量分数为99%)、N-甲基-4-甲氧基苯胺(质量分数为98%)、2-(7-氮杂苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯[2-(7-azabenzotriazol-1-yl)-N,N,N',N'-tetramethyluronium hexafluorophosphate,HATU,质量分数为98%],均购自上海泰坦科技有限公司;无水硫酸钠、氯化钠、氯化铵、柠檬酸、碳酸氢钠、氢氧化钠、三氟甲苯、无水乙醇、N,N-二甲基乙酰胺、二氯甲烷、乙酸乙酯、石油醚均为分析纯,均购自新疆安谱实验器材有限公司;GF254 硅胶板和柱层析硅胶(200 目,青岛海洋化工有限公司)。
2 方法与结果
2.1 化合物4a 和4b 的计算机辅助设计
本文所选用的复合晶体结构数据均来源于Protein Data Bank(www. rcsb. org),编号分别为6BR1和5YZ3。蛋白结构准备和分子对接所使用的软件为Discovery Studio 3.0(DS3.0)。
2.1.1 蛋白结构6BR1 和5YZ3 的叠合 蛋白结构6BR1 和5YZ3 的预处理:保留晶体结构中1 个α、β二聚体及配体小分子和金属离子,并保留6BR1 中的水分子675,将其余部分删除。用Prepare Protein模块进行蛋白结构准备,参数设置为默认值。用Molecular Overlay 模块对准备好的蛋白进行一致性叠合,立体因素和静电因素各按50%概率计算[15]。叠合结果显示,二者相似度为0.946,化合物2 中的喹唑啉环和化合物3 中的苯环有一定程度的重叠,而四氢喹啉环和吡咯并[2,3-d]噻唑分别占据不同的结合位点,尤其是吡咯并[2,3-d]噻唑5-甲酰胺基团与βN165、βE198 和βV236 存在氢键相互作用(见图2),表明在喹唑啉环2 位引入吡咯并[2,3-d]噻唑甲酰胺基团可能会增加其与微管蛋白的相互作用;同时考虑到化合物2 中的喹唑啉环与化合物3 中的苯环在走向上有一定的差异,因此在喹唑啉环和酰胺键之间引入了亚甲基或1,2-亚乙基形成一定的自由度,且化合物1 中的N-甲基-4-甲氧基苯环较化合物2 中的四氢喹啉环可能更有利于化合物与蛋白的诱导契合且易于合成,由此设计了化合物4a 和4b,并进行了分子对接。

图2 化合物2 与微管蛋白结合位点(PDB:6BR1)和化合物3与微管蛋白结合位点(PDB:5YZ3)叠合示意图Fig.2 Superimposition of the tubulin-2 (PDB:6BR1) and the tubulin-3 (PDB:5YZ3) complex structures
2.1.2 化合物4a 和4b 与蛋白结构5YZ3 的分子对接 以2.1.1 中准备好的5YZ3 为对接蛋白,结合位点是以化合物3 为中心、10Å 为半径的球形区域。使用Full Minimization 模块对配体小分子进行了能量最小化,并添加了Chemistry at Harvard Macromolecular Mechamics (CHARMM)力场。用CDOCKER 模块进行了分子对接,参数设置为默认值,主要包括随机构象产生、构象优化和模拟退火等。用In Situ Ligand Minimization 对CDOCKER 对接结果进行了能量最小化,球形区域内的氨基酸残基设定为柔性。对接结果 用 Generalized Born with Molecular Volume(GBMV)溶剂模型计算结合能[15]。
首先用化合物3 验证对接方法,结果显示,最优构象与复合晶体结构中配体构象的走向基本一致,RMSD 值为0.989 Å,结合能为-31.17 kcal·mol-1,表明对接方法效果较好。在化合物4a 和4b 的对接结果中,化合物4a 与化合物3 结构中的吡咯并[2,3-d]噻唑环有较好的重叠,分别与氨基酸残基βN165 和βV236 形成1 个氢键,但未像化合物3 还与βE198 形成氢键(见图3);而化合物4b 由于连接链过长导致吡咯并[2,3-d]噻唑-5-甲酰胺基无法与微管蛋白形成有效相互作用。化合物4a 与蛋白的结合能为-27.93 kcal·mol-1。

图3 4a 与微管蛋白(PDB 编码:5YZ3)的分子对接示意图Fig.3 Predicted binding mode of 4a with tubulin protein (PDB code:5YZ3)
2.2 化学合成
本实验首先合成了化合物8,较文献方法[16]在第一步用乙醇钠溶液代替金属钠,操作更方便且提高了收率。对化合物8 与13 的脱Boc 产物进行缩合,得到目标产物4。在关键中间体13 的合成中,发现在氢氧化钠的作用下,在室温下即可由化合物11 生成化合物12,化合物12 与对甲苯磺酰氯(TsCl)原位生成对甲苯磺酸酯后,直接加入N-甲基-4-甲氧基苯胺反应即得化合物13。
2.2.1 合成路线 化合物4 的合成路线见图4。

图4 化合物4 的合成路线Fig.4 Synthetic route of compounds 4
2.2.2 化合物的合成及表征
2.2.2.1 化合物7 室温下,将5-噻唑甲醛(564 μL,6.5 mmol)和叠氮乙酸乙酯(3.0 mL,26 mmol)溶于30 mL 无水乙醇中,在N2保护下冷却到-10 ℃,滴加乙醇钠(11.2 mL,32.5 mmol)至10 mL 无水乙醇溶液中,滴完后在0 ℃下搅拌5 h。将反应物倒入200 mL碎冰和250 mL 饱和氯化铵溶液中,析出固体物,抽滤,干燥,得到黄色固体物861 mg,即2-叠氮基-3-(5-噻唑基)丙烯酸乙酯,收率为59%。室温下,将2-叠氮基-3-(5-噻唑基)丙烯酸乙酯(580 mg,2.59 mmol)溶于5 mL 干燥三氟甲苯中,在N2保护下滴加到10 mL沸腾的三氟甲苯中,滴完后回流4 h。减压除去三氟甲苯,得到白色固体物442 mg,即化合物7。收率为87%;熔点为119~120 ℃;1H-NMR (400 MHz,CDCl3)δ(ppm) 10.80 (brs,1H),8.80 (s,1H),7.14(d,J=1.9 Hz,1H),4.41 (q,J=7.1 Hz,2H),1.41 (t,J=7.1 Hz,3H);HR-ESI-MSm/z197.038 5 [M+H]+(计算值为197.037 9,C8H9N2O2S)。
2.2.2.2 化合物8 将4H-吡咯[2,3-d]噻唑-5-羧酸乙酯(7,345 mg,1.76 mmol)悬浮于4 mL 四氢呋喃与8 mL 水的混合溶剂中,加入氢氧化锂(337 mg,14.1 mmol),室温下搅拌12 h。将反应物倒入约30 mL冰水中,用2 mol·L-1盐酸调节pH 值至2,用乙酸乙酯萃取3次,每次30 mL,合并有机相,用饱和氯化钠溶液洗,加入无水硫酸钠干燥过夜,减压除去乙酸乙酯,得到浅粉色固体285 mg,即化合物8。收率为96%;熔点为230~234 ℃( 碳化);1H-NMR (600 MHz,d-DMSO)δ(ppm)δ12.66 (s,1H),9.03(s,1H),7.08(d,J=1.9 Hz,1H);HR-ESI-MSm/z169.007 0[M+H]+(计算值为169.006 6,C6H5N2O2S)。
2.2.2.3 化合物11a 室温下,将N-Boc-甘氨酸(1.92 g,11 mmol)溶于30 mL 二氯甲烷中,依次加入2-氨基苯甲酰胺(1.36 g,10 mmol)和N,N-二异丙基乙基胺(3.5 mL,20 mmol),冰浴冷却至0 ℃后加入EDCI·HCl(2.88 g,15 mmol)和HOBt(2.03 g,15 mmol),继续在该温度下搅拌30 min 后,升温至室温,反应24 h。向反应物中加入100 mL 二氯甲烷,分别用50 mL 质量浓度为50 g·L-1的柠檬酸溶液、质量浓度为50 g·L-1的碳酸氢钠溶液和饱和食盐水洗,水相再用50 mL 二氯甲烷依次萃取1 遍,合并有机相,加入无水硫酸钠干燥过夜,减压除去乙酸乙酯,得到白色固体2.66 g,即化合物11a。收率为82%;熔点为150~153 ℃;HR-ESI-MSm/z294.144 7 [M+H]+(计算值为294.144 8,C14H20N3O4)。
2.2.2.4 化合物11b 操作同11a。使用试剂NBoc-β-丙氨酸(2.08 g,11 mmol),2-氨基苯甲酰胺(1.36 g,10 mmol),N,N-二异丙基乙基胺(3.5 mL,20 mmol),EDCI·HCl(2.49 g,13 mmol),HOBt(1.49 g,11 mmol)反应,得到白色固体2.82 g,即化合物11b。收率为83%;熔点为158~161 ℃;HRESI-MSm/z330.143 1[M+Na]+( 计算值为330.142 4,C15H21N3NaO4)。
2.2.2.5 化合物12a 室温下,将2-(2-N-叔丁氧羰基氨基乙酰氨基)苯甲酰胺(11a,2.15 g,7.33 mmol)溶于10 mL 乙醇中,加入10 mL 质量浓度为50 g·L-1的氢氧化钠溶液,室温下搅拌10 h。将反应物倒入约100 mL 冰水中,用2 mol·L-1盐酸调节pH 值至7,析出白色沉淀物,抽滤,干燥,得到白色固体1.85 g,即化合物12a。收率为92%;熔点为205~207 ℃;1HNMR (600 MHz,DMSO) δ 12.13 (s,1H),8.09 (d,J=7.2 Hz,1H),7.82~7.76 (m,1H),7.60(d,J=8.0 Hz,1H),7.48(t,J=7.2 Hz,1H),7.17(t,J=5.4 Hz,1H),4.09(d,J=6.0 Hz,2H),1.40(s,9H);HR-ESI-MSm/z276.134 2 [M+H]+(计算值为276.134 3,C14H18N3O3)。
2.2.2.6 化合物12b 操作同12a。使用试剂2-(3-N-叔丁氧羰基氨基丙酰氨基)苯甲酰胺(11b,0.85 g,2.77 mmol)反应得到白色固体0.74 g,即化合物12b。收率为92%;熔点为146~149 ℃。直接用于下一步反应。
2.2.2.7 化合物13a 室温下,将2-(N-叔丁氧羰基氨基甲基)-4-羟基喹唑啉(12a,1.73 g,6.28 mmol)悬浮于60 mL 二氯甲烷中,依次加入三乙胺(1.76 mL,12.6 mmol)、N,N-二甲基-4-氨基吡啶(77 mg,0.63 mmol)和对甲苯磺酰氯(1.26 g,6.61 mmol),室温下搅拌12 h。再向反应物中加入N-甲基-4-甲氧基苯胺(0.95 g,6.93 mmol),室温下继续搅拌12 h。将反应物倒入约50 mL 冰水中,用乙酸乙酯萃取3次,每次50 mL,合并有机相,用饱和氯化钠溶液洗,加入无水硫酸钠干燥过夜,减压除去乙酸乙酯,粗品经柱色谱分离(乙酸乙酯∶石油醚=10%~20%),得到淡黄色固体1.61 g,即化合物13a。收率为65%;熔点为118~120 ℃;1H-NMR (600 MHz,d-DMSO)δ(ppm)δ7.68(d,J=8.4 Hz,1H),7.61(t,J=7.5 Hz,1H),7.21(d,J=8.4 Hz,2H),7.13~7.06(m,2H),7.00(d,J=9.0 Hz,2H),6.95(d,J=8.4 Hz,1H),4.29(d,J=6.0 Hz,2H),3.78(s,3H),3.52(s,3H),1.43(s,9H);HR-ESI-MSm/z395.207 6 [M+H]+(计算值为395.207 8,C22H27N4O3)。
2.2.2.8 化合物13b 操作同13a。使用试剂2-(N-叔丁氧羰基-2-氨基乙基)-4-羟基喹唑啉(12b,0.43 g,1.5 mmol)、三乙胺(0.56 mL,4.0 mmol)、N,N-二甲基-4-氨基吡啶(24 mg,0.2 mmol)和对甲苯磺酰氯(0.37 g,1.6 mmol)、N-甲基-4-甲氧基苯胺(0.22 g,1.6 mmol)反应得到淡黄色固体0.38 g,即化合物13b。收率为61%;熔点为106~108 ℃;1H-NMR(600 MHz,d-DMSO)δ(ppm)7.67(d,J=7.8 Hz,1H),7.59(t,J=7.8 Hz,1H),7.20(d,J=9.0 Hz,2H),7.07(t,J=7.2 Hz,1H),6.98(d,J=9.0 Hz,2H),6.96(d,J=8.4 Hz,1H),6.88(t,J=5.5 Hz,1H),3.78(s,3H),3.47~3.50(m,5H),2.95(t,J=7.3 Hz,2H),1.36(s,9H);HR-ESI-MSm/z409.223 9 [M+H]+(计算值为409.223 4,C23H29N4O3)。
2.2.2.9 化合物4a 将2-(N-叔丁氧羰基氨基甲基)-4-(N-甲基-4-甲氧基苯胺基)喹唑啉(13a,0.33 g,0.84 mmol)溶于5 mL 乙酸乙酯中,加入0.5 mL浓盐酸,升温至60 ℃搅拌6 h。向反应物中分别加入30 mL 乙酸乙酯和30 mL 水,分出水相,用质量浓度为50 g·L-1的氢氧化钠溶液调节pH 值至8,用乙酸乙酯萃取3 次,每次50 mL,合并有机相,用饱和氯化钠溶液洗涤,加入无水硫酸钠干燥过夜,减压除去乙酸乙酯,得到褐色固体0.18 g,即化合物2-氨甲基-4-(N-甲基-4-甲氧基苯胺基)喹唑啉,收率为73%。将4H-吡咯[2,3-d]噻唑-5-甲酸(8,84 mg,0.5 mmol)溶于5 mLN,N-二甲基甲酰胺中,依次加入N,N-二异丙基乙基胺(0.22 mL,1.25 mmol)和HATU(285 mg,0.75 mmol),室温下搅拌30 min,加入制备的2-氨甲基-4-(N-甲基-4-甲氧基苯胺基)喹唑啉(148 mg,0.5 mmol),继续在室温下搅拌24 h。将反应物倒入约50 mL 冰水中,析出固体物,抽滤,干燥,粗品经柱色谱分离(甲醇-二氯甲烷,甲醇2.5%~5.0%),得到白色固体49 mg,即化合物4a。收率为22%;熔点为215~218 ℃;1H-NMR(600 MHz,d-DMSO)δ(ppm)12.49 (s,1H),8.95(s,1H),8.77(t,J=5.8 Hz,1H),7.69(d,J=7.6 Hz,1H),7.63~7.58(m,1H),7.25(t,J=3.6 Hz,1H),7.23~7.18(m,2H),7.08(ddd,J=8.3,6.9,1.2 Hz,1H),7.01~6.97(m,2H),6.97~6.93(m,1H),4.65(d,J=5.9 Hz,2H),3.79(d,J=6.8 Hz,3H),3.49(s,3H);13C-NMR(150 MHz,DMSO)δ162.11,160.99,160.80,157.77,154.54,152.95,141.01,140.53,132.07,131.61,127.73,127.30(2*C),125.76,124.44,115.32(2*C),114.68,113.82,101.23,55.37,44.96,42.37;HR-ESI-MSm/z445.144 3 [M+H]+(计算值为445.144 1,C23H21N6O2S)。
2.2.2.10 化合物4b 操作同4a。2-(N-叔丁氧羰基-2-氨基乙基)-4-(N-甲基-4-甲氧基苯胺基)喹唑啉(13b,0.41 g,1.0 mmol)经脱Boc 保护得到褐色固体0.24 g,即化合物2-(2-氨基乙基)-4-(N-甲基-4-甲氧基苯胺基)喹唑啉,收率为78%。使用4H-吡咯[2,3-d]噻唑-5-甲酸(8,65 mg,0.39 mmol)、N,N-二异丙基乙基胺(0.17 mL,0.98 mmol)、HATU(225 mg,0.59 mmol)、2-(2-氨基乙基)-4-(N-甲基-4-甲氧基苯胺基)喹唑啉(120 mg,0.39 mmol)反应得到白色固体56 mg,即化合物4b。收率为31%;熔点为194~196 ℃;1H-NMR(400 MHz,d-DMSO)δ(ppm)12.39(s,1H),8.91(s,1H),8.34(s,1H),7.68(d,J=7.6 Hz,2H),7.61~7.57(m,1H),7.19(d,J=8.8 Hz,2H),7.10~7.03 (m,2H),6.98(d,J=8.8 Hz,2H),3.92~3.71(m,5H),3.49(s,3H),3.12(t,J=7.2 Hz,2H);13C-NMR(100 MHz,d-DMSO)δ163.30,161.05,160.59,157.63,152.82,140.94,131.87,131.72,131.58,130.64,128.65,127.74,127.24(2*C),125.68,124.24,115.26(2*C),114.54,100.84,55.35,42.35,38.08,37.78;HRESI-MSm/z459.159 2 [M+H]+( 计算值为459.159 8,C24H23N6O2S)。
2.3 A549 肿瘤细胞增殖抑制活性的筛选
本实验用磺酰罗丹明B(sulforhodamine B,SRB)法[17-19]。取处于对数生长期的人肺癌A549 细胞接种于96 孔板中,密度约为1×105个·孔-1。37 ℃下培养24 h 后弃去上清液,空白组和对照组各加入100 μL 不含药物的培养基,实验组分别加入含阳性药、化合物4a 和4b 的培养基,每组设置5 个复孔。继续培养48 h后,弃去上清液,用100 mL·L-1三氯乙酸在4 ℃下固定1 h,用纯净水洗涤5 次,风干。将存活的细胞在室温下用4 g·L-1SRB 染色20 min,用10 mL·L-1乙酸洗涤5 次。将结合的SRB 溶解于10 mmol·L-1的Tris(缓冲液)中,用酶标仪测定540 nm 处的吸光度值,计算细胞生长抑制率。根据药物浓度与抑制率作图,计算GI50值,实验平行3 次。阳性药为紫杉醇。
经测定,化合物4a 和4b 均有一定的体外肿瘤细胞增殖抑制活性,化合物4a 的活性强于4b,但均弱于紫杉醇。结果见表1。

表1 化合物4a、4b 抑制A549 细胞增殖的GI50值和抑制微管聚集的IC50值Tab.1 GI50 value against A549 and the IC50 value against tubulin polymerization of compounds 4a and 4b
2.4 体外微管蛋白聚集抑制活性的筛选
用循环离心法制备的猪脑微管蛋白进行活性测试[20-21]。将不同浓度的药物与微管蛋白孵化后,在冰浴下加入GDP,转移到0 ℃的比色杯中,随后升温至37 ℃,测定该过程中340 nm 处的吸光度值。与对照组比较,计算不同药物浓度下的抑制率。根据药物浓度与抑制率作图,计算IC50值,实验平行3 次。阳性药为CA-4。
实验结果显示,4a 表现出了较弱的微管聚集抑制活性,但明显弱于CA-4,4b 并未表现出明显的微管聚集抑制活性。结果见表1。
3 讨论
本实验通过将化合物2、3 与微管蛋白的复合晶体结构进行叠合,利用“拼接”策略设计了化合物4a和4b,在合成4H-吡咯[2,3-d]噻唑-5-羧酸的基础上,通过酰胺缩合、分子内成环、亲核取代和酰胺缩合4 步反应合成了目标化合物,总收率分别为7.9%、11.3%,目标化合物结构经1H-NMR、13C-NMR 和HR-ESI-MS 确证。
文献报道的作用于秋水仙碱结合位点的化合物的微管聚集抑制活性与细胞增殖抑制活性一般相差上百倍,本文设计的化合物4a 和4b 在微管聚集抑制活性和细胞增殖抑制活性方面也表现出类似的特点,表明微管蛋白仍可能是化合物的主要作用靶点。化合物4a 的微管聚集抑制活性优于4b,与分子对接的结果基本一致,表明计算机辅助设计结果具有一定的参考价值。但化合物4a 未表现出预期较化合物2、化合物3 更强的微管聚集抑制活性,可能与化合物分子结构中喹唑啉环和吡咯并[2,3-d]噻唑环间存在一定的刚性或连接链长度不合适有关,导致化合物4a 无法同时与化合物2、化合物3 相作用的所有氨基酸残基形成相互作用。
尽管化合物4a 与化合物1、化合物2 相比肿瘤细胞增殖和微管聚集抑制活性差异较大,但肿瘤细胞增殖抑制活性却仅略弱于化合物3 的活性更好的衍生物14(见图5,A549 细胞IC50值为40 μmol·L-1)[12]。LI Y 等[22]报道了一类咔啉类化合物,其代表化合物15(见图5)的结合位点与化合物3 基本重叠,机制研究发现化合物15 具有“分子胶水”的特征,能特异性诱导降解微管蛋白β亚基,在卵巢癌异种移植裸鼠模型中表现出良好的肿瘤抑制活性,且无明显的不良反应,表明可将化合物4a 作为先导化合物,进行深入的构效关系研究并开展类似的机制研究,得到具有临床开发潜力的化合物。
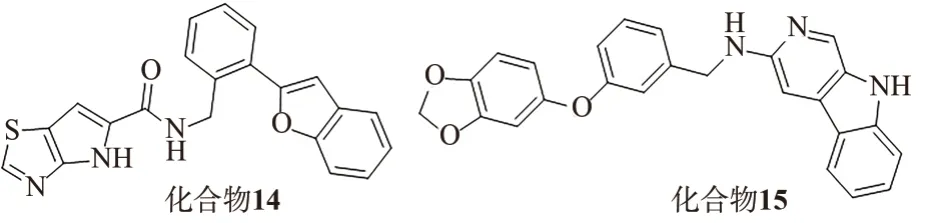
图5 化合物14 和15 的结构Fig.5 Structure of compounds 14 and 15
综上所述,本研究通过药物拼接的方法得到了一类新结构类型的verubulin 衍生物,其中化合物4a预期可占据微管蛋白秋水仙碱结合位点更广泛的区域,这种基于复合晶体结构的分子“拼接”方法可为发现新型微管聚集抑制剂提供借鉴。
猜你喜欢
黑龙江大学自然科学学报(2022年4期)2022-11-17
——水芹主要害虫识别与为害症状
长江蔬菜(2022年13期)2022-07-29
云南化工(2021年7期)2021-12-21
四川轻化工大学学报(自然科学版)(2021年3期)2021-08-30
中外医疗(2016年15期)2016-12-01
合成化学(2015年10期)2016-01-17
化工进展(2015年6期)2015-11-13
郑州大学学报(工学版)(2015年1期)2015-03-24
华东师范大学学报(自然科学版)(2014年4期)2014-03-11
无机化学学报(2014年4期)2014-02-28
