
口服抗高血压药物研究进展
2024-01-23 01:30马科星张潇惠新平武全香
大学化学 2023年12期
马科星,张潇,惠新平,*,武全香,*
1 兰州大学化学化工学院,兰州 730000
2 兰州大学公共卫生学院,兰州 730000
高血压是指以体循环动脉血压增高(收缩压≥140 mmHg和/或舒张压≥90 mmHg)为主要特征,可伴有心、脑、肾等器官功能或器质性损害的临床综合征,是严重危害居民健康的慢性疾病。高血压患者中80%非高危人群可通过控制饮食、运动、戒烟、戒酒、减重等降低血压,20%高危人群除了调整生活方式还需要进行药物治疗。我国现有高血压患者约2.45亿,抗高血压药物需求快速增长[1]。
2022年11月13日,《中国高血压临床实践指南》发布,推荐我国成人高血压诊断界值下调为收缩压≥130 mmHg和/或舒张压≥80 mmHg。研究结果表明,血压水平130–139/80–89 mmHg使心血管发病及死亡的相对风险升高约39%–90%,11%冠心病可归因于该血压水平。此外,我国40岁以下青年人群中,血压水平130–139/80–89 mmHg的人群脑卒中风险、特别是脑出血风险明显增加。因此,新标准对我国心脑血管疾病,特别是脑卒中的预防具有必要性和可行性[2]。
抗高血压药物主要分为血管紧张素I转化酶抑制剂(angiotensin converting enzyme inhibitors,ACEI)、血管紧张素II受体阻断剂、血管扩张药、利尿降压药、β受体阻断药、钙拮抗药和交感神经抑制药七类。美国食品药品监督管理局(Food and Drug Administration,FDA)批准上市的抗高血压药物数量统计显示(图1),抗高血压药物研发从20世纪60年代开始,20世纪末进入高峰期。进入21世纪,由于新药开发成本增加、临床获利下降,高血压基础医学研究达到瓶颈期,抗高血压药物研发大幅减缓。高血压治疗最直接有效的方式是降低血压,药物研发主要集中于降压药物[3]。目前,临床使用的抗高血压药物主要是基于肾素-血管紧张素-醛固酮系统(renin-angiotensin-aldosterone system,RAAS)的靶向药物(图2)[4]。本文对2011年以来FDA批准上市的血管扩张药、β受体阻断药、血管紧张素II受体拮抗剂类抗高血压药物研究进展进行了总结。
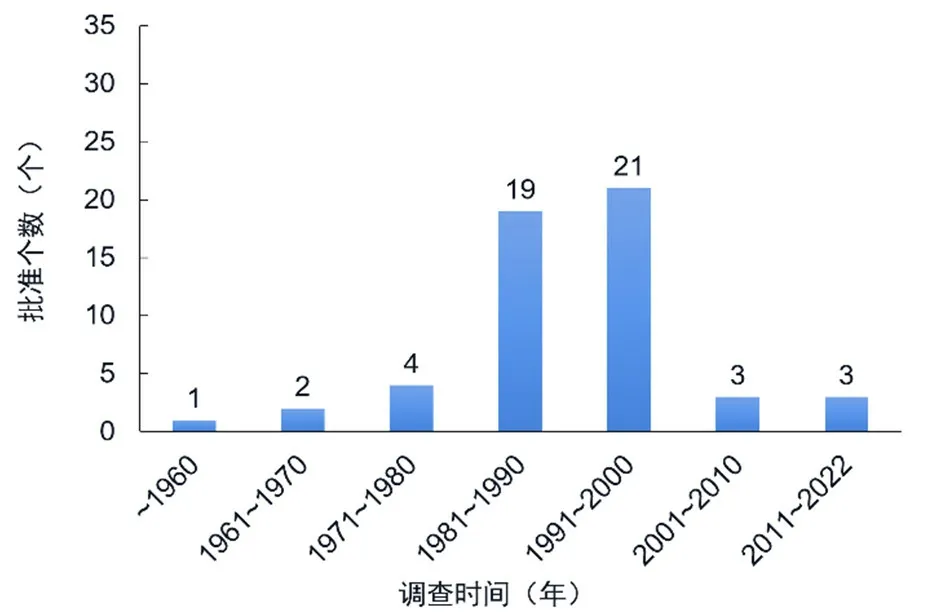
图1 FDA批准上市的抗高血压药物数量

图2 肾素-血管紧张素-醛固酮系统的经典靶点
1 血管扩张药——赛乐西帕
能直接扩张小血管平滑肌或通过作用于肾上腺素受体而舒张血管的药物即为血管扩张药。随着药物的使用,人体逐渐产生耐药性,传统血管扩张药的作用愈发下降。开发新型、高耐药性与普受性的血管扩张药显得尤为重要。2015年12月21日,美国FDA批准爱可泰隆(actelion)公司研发的新药——赛乐西帕(selexipag,商品名Uptravi,1,图3)上市。赛乐西帕是一种口服、高选择性、长效前列环素[prostacyclin(prostaglandin I2),PGI2,2]受体激动剂前药,其作用机理与PGI2一致,具有抗血小板聚集、抑制炎性介质释放和舒张血管作用[5]。
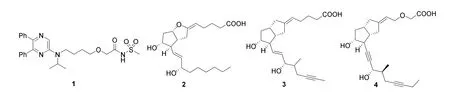
图3 赛乐西帕及前列环素系列药物分子结构
1.1 赛乐西帕先导化合物的发现及结构修饰
1976年,Moncade等[6]从兔子和猪动脉中分离得到了PGI2,PGI2是由血管内皮细胞合成并释放入血的血管活性物质,用于治疗肺动脉高血压(pulmonary arterial hypertension,PAH)。PGI2的不稳定性限制了其临床应用,但以其作为先导化合物,进行结构修饰得到了一系列PGI2类似物,它们能更好地诱导肺血管阻力迅速下降、增加心输出量、降低血压。如何开发稳定、靶向精准的PAH的PGI2受体(prostaglandin I2receptor,IP受体)药物,开启了非前列腺素类IP激动剂药物的研发热潮[7]。
1.1.1 前列环素类
Schering AG公司通过对PGI2侧链修饰得到了血小板活化抑制剂——伊洛前列素(iloprost,3,图3)和西卡前列素(cicaprost,4,图3)[8]。上市的PGI2药物结构与PGI2类似,存在PGI2固有的稳定性差、口服难消化的问题,多以静脉注射给药。
1.1.2 吡唑类修饰物
Meanwell等[9]利用小分子化合物库随机筛选,发现降血脂药辛米贝特(octimibate,5,图4)具有PGI2受体部分激动作用。为了摆脱PGI2的固有缺陷,他们将5中的咪唑环替换为吡唑环,同时改变羧酸链长合成了化合物6(图4),消除了原药物易于水解的性质,但抑制血小板聚集作用弱于化合物2、3和5。
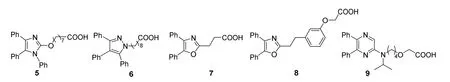
图4 基于辛米贝特修饰的化合物结构
1.1.3 噁唑类修饰物
Meanwell等[9]参考抗炎药奥沙普秦(oxaprozin,7,图4)的结构,将化合物6中的吡唑替换为噁唑,合成了4,5-二苯基噁唑-2-壬酸。利用生物电子等排原理,将亚甲基替换为间氧取代苄基得到了化合物8(图4),其抑制二膦酸腺苷(adenosine diphosphate,ADP)诱导血小板聚集的IC50为0.18 μmol∙L-1,与血小板的竞争结合强度为化合物3的6倍[10]。
1.1.4 二苯吡嗪类修饰物
基于杂环变换策略,Clozel等[11]用吡嗪替换噁唑,通过烷基链长度调节母核与羧基间的距离得到一系列二苯基吡嗪酸衍生物,活性筛选发现化合物9(图4)的活性最强,抑制血小板聚集的IC50为0.2 μmol∙L-1。Kuwano等[12]对其进行了修饰,得到了N-甲磺酰化产物1(赛乐西帕,图3)。1体外结合并激动PGI2受体作用比9降低了13倍,抑制血小板聚集活性降低了26倍,但1体内可水解生成9。多次给药,1不会引起高血压大鼠的快速耐受性。赛乐西帕因其长效、高选择性而批准上市。
1.2 赛乐西帕的构效关系
根据以上研究,构效关系总结如图5所示,母核为二苯吡嗪酸系列衍生物呈现出强抑制活性,优于二苯基吡咯、噻唑、吡唑、三唑、吡嗪、三嗪、嘧啶酮、苯并吡嗪酮或苯并嘧啶二酮等烷基羧酸。羧基与母核的距离以C8为最适长度,环的不同位置分别去除一个苯环均使活性降低。将连接吡嗪环与碳链的-CH2-作电子等排变换,用-O-、-S-、N-Me、亚磺酰或磺酰基置换,其中N-Me替代活性最优。进而对N-Me的甲基作变换,合成的N-烷基或环烷基化合物中氮原子连接苄基等大取代基不利于活性,甲基、乙基、环丙基和异丙基等小尺寸烷基都显示高活性,其中N-异丙基化合物不仅活性高,而且选择性强。继续将羧基β位引入氧原子,可避免羧酸的β氧化,提高体内代谢稳定性。前药化合物中含有游离羧基,极性较强不利于吸收,为了降低羧基的离解性,将其替换为酰氨的N-甲磺酰化合物,在体内可恢复生成羧基[13]。
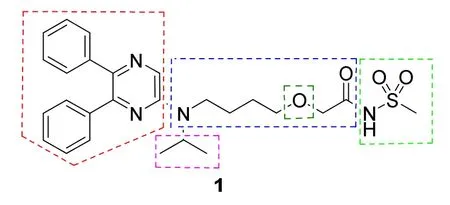
图5 赛乐西帕的结构活性关系图
1.3 赛乐西帕的合成
赛乐西帕最早由Asaki等[14]合成,但成本较高且条件较苛刻,不利于工业化生产。随后,尚振华等[15]对其进行了改进。以1,2-二羰基化合物10和甘氨酰胺盐酸盐为原料,经环化、氯代得到12,12先后与N-异丙基丁-1-醇和N-(2-氯乙酰基)甲基磺酰胺反应得到了赛乐西帕(1)。该路线缩短了反应步骤、提高了收率,产物易提纯,适用于工业化生产(图6)。

图6 赛乐西帕合成路线
2 β受体阻滞剂类药——琥珀酸美托洛尔
1962年,帝国化学工业公司Black等[16]发现了β受体阻滞剂,是β肾上腺素能受体(adrenergic receptors,ARs)拮抗剂。早期的β受体阻滞剂选择性不高,第三代β受体阻滞剂提高了选择性,还具有血管舒张特性。2018年1月26日,美国FDA批准上市的β受体阻滞剂——琥珀酸美托洛尔(metoprolol succinate,13,图7)是一种高选择性的口服药物,可用于治疗高血压、冠心病、慢性心力衰竭和心律失常等[17]。
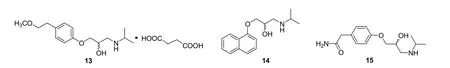
图7 β受体阻滞剂类药物分子结构
2.1 美托洛尔先导化合物的发现、结构修饰与构效关系
以第一代β受体阻滞剂普萘洛尔(propranolol,14,图7)为先导,帝国化学工业公司研发了第二代β受体阻滞剂阿替洛尔(atenolol,15,图7),1981年FDA批准上市,这是FDA批准的第一个β受体阻滞剂药物。结构修饰发现,14中与萘环相连的氧原子被其他原子取代降压效果降低,萘环可被其他不饱和环取代,环上可以含有甲基、硝基等取代基,萘环2,4-或2,3,6-取代时活性最好,侧链末端氮原子上必须是叔丁基或异丙基,若其他基团取代活性下降,手性碳为S构型时活性最强[18]。基于构效关系研究,Roche公司和AHP公司分别研发了卡维地洛(carvedilol,16,图8)和萘比洛尔(nabibiolol,17,图8),瑞典研发了美托洛尔(metoprolol,18,图8)等[19,20]。18生物兼容性低、药效时间短且存在副作用,Sandberg等[21]将美托洛尔分别与酒石酸、琥珀酸作用,得到了酒石酸美托洛尔(metoprolo tartrate,1992年FDA批准上市)和琥珀酸美托洛尔(13)。
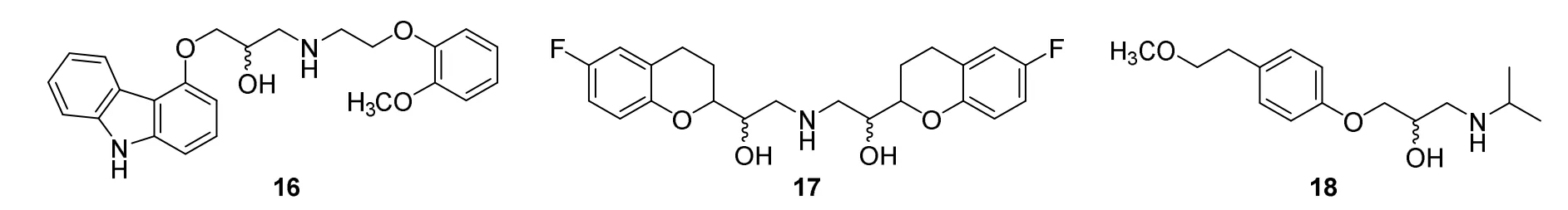
图8 以14为前体修饰的β受体阻滞剂药物分子结构
2.2 琥珀酸美托洛尔的合成
美托洛尔(18)最早由Gurjar等[22]合成,但催化剂OsO4价格昂贵,毒性高,难以实现大规模生产。牟佳佳等[23]以化合物19和环氧氯丙烷为原料,在溴化四丁基铵作用下发生亲核取代反应得到20,再在AlCl3催化下使用异丙胺开环得到美托洛尔,最后与琥珀酸的饱和乙醇溶液反应得到琥珀酸美托洛尔(13)。该方法步骤简单,收率较高且绿色环保(图9)。

图9 琥珀酸美托洛尔的合成路线
3 血管紧张素II受体拮抗剂——阿齐沙坦酯
血管紧张素(angiotensin,AT)II是肾素-血管紧张素-醛固酮系统重要的升血压肽,血管紧张素II受体有AT1–AT4亚型[24]。目前临床使用或开发中的“沙坦”类抗高血压药都是选择性地阻断AT1[25]。2011年2月25日,美国FDA批准日本武田制药公司(Takeda)研发的AII受体阻滞剂阿齐沙坦酯(Azilsartan Medoxomil,商品名Edarbi,21,图10)用于成人高血压治疗。与缬沙坦(valsartan,22)和奥美沙坦(omesartan,23)相比,阿齐沙坦酯24小时内的降压效果更好[7,8]。
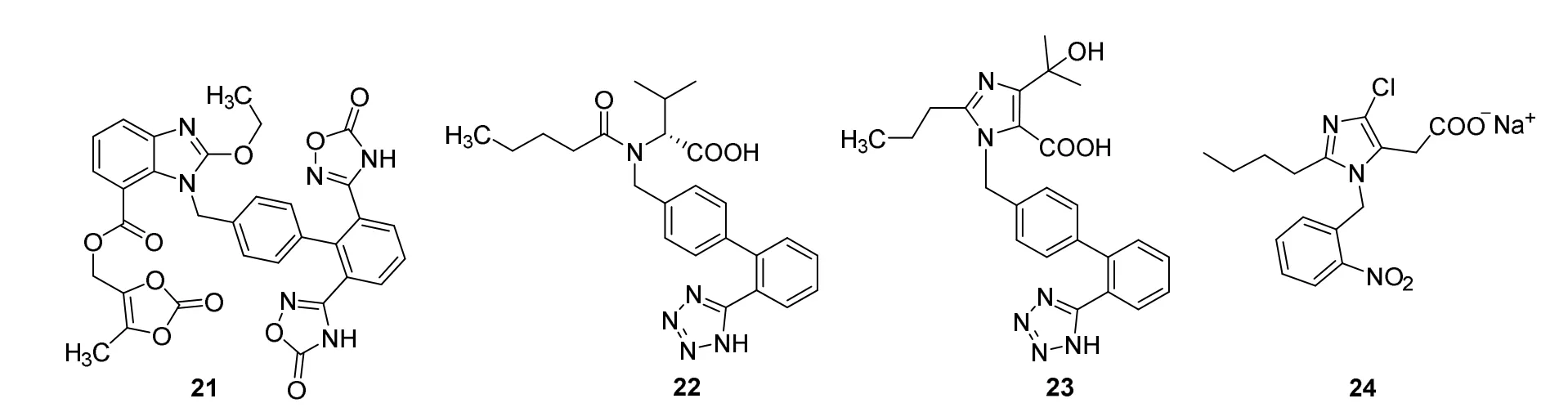
图10 沙坦类药物及先导化合物结构
3.1 阿齐沙坦酯先导化合物的发现及结构修饰、构效关系
1982年,研究发现以化合物24(图10)为代表的咪唑5-乙酸类似物具有抗高血压作用,24能特异性阻断AII受体。尽管其拮抗活性较弱,但不具有激动剂活性。研究人员通过计算机分子叠合法模型揭示了24与AII受体存在三个共同的结构特征:离子化羧基、咪唑环和烃基侧链。Dupont Merck Pharmaceutical公司[26]通过对24结构改造,得到了第一代AII受体拮抗剂——氯沙坦(Losartan,25,图11),可阻断血管紧张素II所致的动脉血管收缩、交感神经兴奋和压力感受器敏感性增加等,达到高效降压效果。基于25的构效关系和结构修饰,得到了一系列沙坦类药物,如缬沙坦、奥美沙坦、厄贝沙坦(irbesartan,26)、坎地沙坦(candesartan,27)和美阿沙坦钾(azilsartan medoxomil potassium,28)等(图11)。
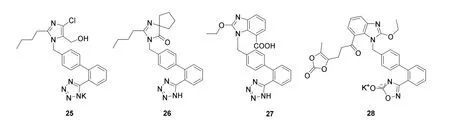
图11 部分沙坦类药物分子结构
目前,国内抗高血压药物虽以钙拮抗剂和ACEI(普利类)占主导,但由于沙坦类药物优良的降压效果,副作用小,患者依从性好,而且有近10年的临床使用和降压作用的肯定性评价,使沙坦类药物有良好市场。鉴于大多ACEI和钙拮抗剂药物专利保护即将失效,原生产抗高血压药物的厂家正致力于沙坦类药物的研发和推广,目前沙坦类在抗高血压药物市场占据了重要位置[27]。许多高血压或充血性心力衰竭患者胰岛素耐药并容易发展为II型糖尿病,沙坦类药物可以预防高风险人群的发病[28]。随着单剂与复方制剂的不断开发及新的适应证的不断发现,沙坦类药物发展前景广阔。
3.2 阿齐沙坦酯的合成
阿齐沙坦酯的合成多以阿齐沙坦为原料,岑均达等[29]以3-硝基邻苯二甲酸-1-甲酯(29)为原料,经Curtius重排得到30,后与2’-氰基-4-溴甲基联苯反应得到31,31在浓盐酸作用下烷氧基甲酰胺基脱除叔丁氧甲酰基、Pd/C催化氢化硝基、两个氨基与原碳酸四乙酯发生环化反应得到34。34在50%羟胺水溶液/三乙胺-乙醇中回流得到35,随后取代得到36,在乙醇中发生分子内氨解得到37,水解得到阿齐沙坦38(图12)。杨和军等[30]以38为原料,酯化后再用盐酸-异丙醇水解得到了21。改进后的工艺反应条件温和,后处理简便,纯度 >99.8%,更适合工业化生产。
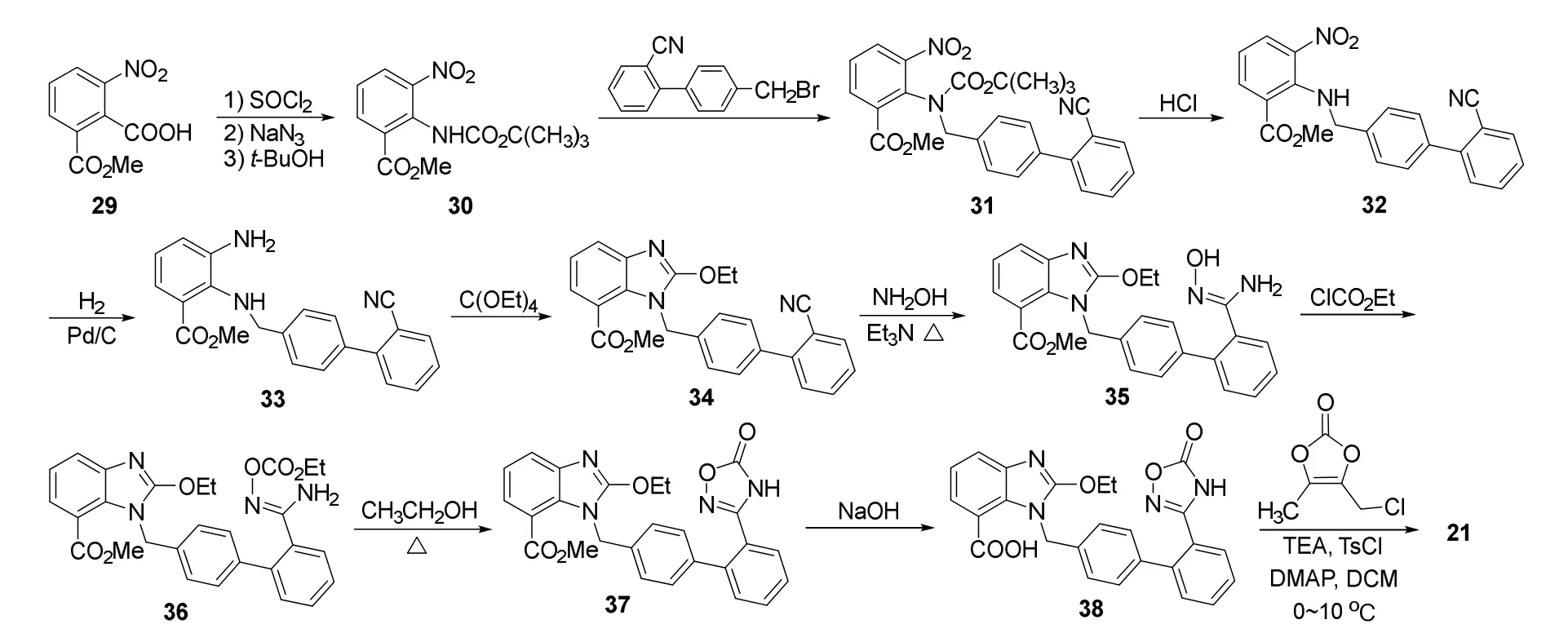
图12 阿齐沙坦酯钾盐的合成路线
4 总结与展望
高血压发病率越来越高,足剂量、长效制剂、联合用药、个体化是治疗高血压的基本原则。《世界卫生组织成人高血压药物治疗指南》《国际高血压学会全球高血压实践指南》和《中国高血压防治指南》等相继发布,指导人们合理用药治疗高血压。传统的降压药物单药疗效欠佳、药物耐受性及患者依从性差等因素导致患者血压达标率不高,不能达到预期治疗效果。目前,抗高血压药物的研发集中在对传统降压药物的改进和探索新作用靶点的降压药物方面。新型抗高血压治疗靶点的发现和药物研发迫在眉睫,相信随着细胞分子生物学,特别是“组”生物学的发展,新靶点、新通路、新机理、新技术的不断出现,必将推动抗高血压药物开发的快速发展。
猜你喜欢
家庭医药(2023年9期)2023-09-16
现代实用医学(2022年10期)2022-12-08
世界科学技术-中医药现代化(2021年12期)2021-04-19
合成化学(2015年2期)2016-01-17
中国卫生标准管理(2015年2期)2016-01-14
中国继续医学教育(2015年1期)2016-01-06
西南医科大学学报(2015年1期)2015-08-22
中国当代医药(2015年26期)2015-03-01
中国当代医药(2015年21期)2015-03-01
西南军医(2015年5期)2015-01-23
