
第55届国际化学奥林匹克试题(理论部分)
2024-01-23 01:30张丽荣郭玉鹏张明涛王颖霞裴坚
大学化学 2023年12期
张丽荣,郭玉鹏,张明涛,王颖霞,裴坚
1 吉林大学化学学院,长春 130012
2 南开大学化学学院,天津 300071
3 北京大学化学与分子工程学院,北京 100871
第1题 分子成像
分子成像是医学诊断的有力工具。作为同位素99gTc(g=基态)的核异构体,99mTc(m=亚稳态)具有良好的辐射特性(γ辐射体,t1/2=6.015 h),可用于成像。99mTc在锝发生器中通过母核的β-衰变得到,以99mTc-高锝酸盐[99mTcO4]-形式存在。
1.1 确定如下核反应中99mTc的母核(A)和放射出的粒子(B):A —99mTc+B
1.2 写出图1-1中所示99mTc-探针中放射性金属的氧化态。

图1-1 a)99mTc-Sestamibi(Cardiolite)心脏成像;b)99mTc-DBODC5心脏成像;c)Neurolite®脑成像;d)骨成像
第七族元素锰(Mn)、锝(Tc)和铼(Re)的氧化还原电势与元素周期表中的总体趋势一致(见图1-2)。
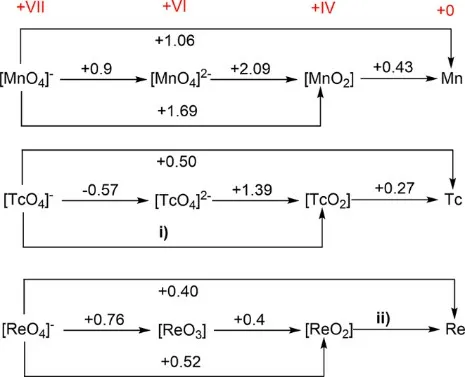
图1-2 酸性条件下锰族三元素的Latimer电势图,以标准氢电极(SHE)为标准,电极电势的单位为伏特(V)
1.3 计算缺失的两个氧化还原电势i)和ii)。
1.4 比较[MnO4]-、[TcO4]-和[ReO4]-,确定哪个是最强的氧化剂。
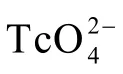
1.6 含有端氧(O=)或氮(N≡)配体且氧化态为+V的Tc和Re(d2组态)的配合物显抗磁性。图1-3给出三种可能的分子轨道能级的示意图。在图中勾选出可以解释所观察到的抗磁现象的轨道能级图并在所选图中画出相应的电子排布。
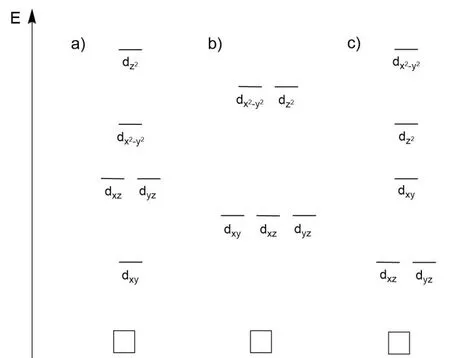
图1-3 三种可能的分子轨道能级的示意图
1.7((C4H9)4N)[99gTcO4]是一种无色粉末。加入浓盐酸后,这种常用于99gTc化学研究的起始化合物会转化为绿色复合物((C4H9)4N)[99gTcOCl4]。写出氧化半反应、还原半反应以及氧化还原总反应。
1.8 临床所用99mTc放射性核素都是采用商业化试剂盒,通过“一锅煮”反应制得。通常情况下,99mTc发生器的洗脱液的活性为12.5 GBq(GBq=109贝克勒尔=每秒衰变109次)。计算这些样品中含有多少摩尔的99mTc(t1/2=6.015 h)。
1.9 在成像标准中,允许注射到患者体内的99mTc约为200 MBq。假设活性不通过排泄物流失。计算患者需要等待多少小时,注射的活性会降低到起始活性的1%以下。
放射性金属的生物共轭是一种化学挑战。最近的一个例子是[99mTcO3(tacn)]+(A)(tacn=1,4,7-三氮杂环壬烷)与烯烃的(3+2)环加成反应。在这里,(3+2)指的是涉及的原子数,而不是电子数。下面的示意图通过标记受保护的碳水化合物,展示了这一反应的实例。

1.10 画出化合物A和B的结构。指出这些化合物中锝的氧化态。
第2题 CO2的电化学还原
近年来,将CO2电化学转化为高附加值产品被认为是缓解大气中CO2含量不断增加所造成的负面气候影响的一种前景广阔、技术可行的方法。为实现这一目标,已经开发了几种技术。其中,通过电化学方法实现CO2还原反应(CO2RR)值得特别关注,因为它能够以可再生能源为动力,将对环境有害的CO2转化为基本化学品。
电催化剂不仅可加速原本缓慢的CO2RR反应,而且也可引导电解反应向所需的反应产物(产物选择性)方向发展。在这种情况下,不仅催化剂化学性质而且其大小和形貌也决定了CO2RR产物的分布。一种设计CO2RR催化剂的新概念基于泡沫型材料的电沉积,这种材料为反应物(如H2O、H2和CO2)提供了大的可接触面积。铜基材料是已知唯一的可催化电解CO2产生大量碳氢化合物和醇类的CO2RR金属催化剂。已知Eo(Cu2+/Cu)=+0.34V,表2-1给出了部分物质的热力学数据。

表2-1 一些物质在标准条件下(T=298.15 K,p=1 bar)的标准生成焓ΔfHϴ和标准熵Sϴ
2.1 写出酸性条件下(i)和(ii)电化学还原过程的半反应方程式并配平:(i)CO2还原成乙醇;(ii)CO2还原成正丙醇。
2.2 将还原过程的半反应与标准氢电极的半反应合并,写出此电池反应式,后者作为阳极。计算CO2还原为乙醇的电池反应的标准电动势。
2.3 泡沫铜电沉积过程在含有0.2 M硫酸铜的1.5 M硫酸水溶液中进行。铜片(1 cm2)和铂箔分别作为阴极和阳极。写出分别发生在阴极和阳极的所有还原和氧化半反应。
图2-1示出析氢反应(HER)中动态氢气泡产生以及金属沉积的原理。图2-2表示分别在不同时间(5 s、20 s和80 s)停止金属沉积后获得的三种泡沫Cu表面的扫描电子显微(SEM)俯视图像。
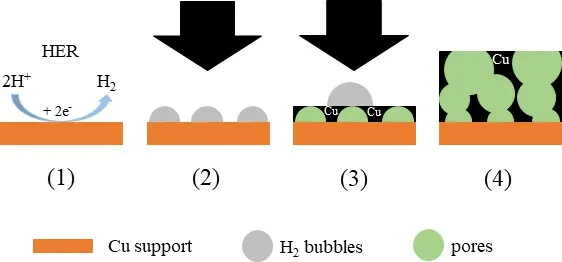
图2-1 泡沫型材料的电沉积示意图
2.4 根据以上机制,在图2-2中标出所示泡沫铜对应的沉积时间(5 s、20 s或80 s,写在图片左上角的空白处)。
双金属铜银体系是以CO2为原料电合成醇类的优良催化剂。在电流密度j=-3 A cm-2(负号表示还原/阴极过程)条件下,保持沉积时间20 s,在铜箔(1 cm2)上电沉积得到5.4 mg双金属铜银泡沫催化剂(90 wt.% Cu,MCu=63.546 g mol-1;10 wt.% Ag,MAg=107.868 g mol-1)。
2.5 计算该金属沉积过程的法拉第效率(FE,表示为%)。FE的定义为(Q产物/Q总)×100%。Q表示电荷。
现在来看一个二氧化碳电解实验:在被二氧化碳饱和的35毫升0.5 M KHCO3电解质溶液中,使用双金属Cu-Ag泡沫为催化剂(90 wt.% Cu;10 wt.% Ag)。电解在j(总)=-30 mA cm-2的恒定电流密度(总)下持续进行3600 s(注意电流密度是根据1 cm2的几何表面积归一化得到的;负号表示还原/阴极过程)。对电解产物进行分析显示,乙醇和正丙醇的质量浓度分别为41.3 mg L-1和7.4 mg L-1。这两种醇均为液态反应产物,在电解反应过程中溶解在电解质中。我们假设在这一过程中形成的唯一副产物是气态氢(H2)。
2.6 计算形成乙醇和正丙醇各需要的电流密度。假定总电流密度不随电解时间变化。(M乙醇=46.08 g mol-1;M正丙醇=60.10 g mol-1)
2.7 计算在298.15 K和1 bar条件下,1 cm2催化剂面积上,形成的氢气体积,假设形成的氢气为理想气体并全部以气态形式放出。(如果你在题2.6中未得到结果,请按照FE(乙醇)=45.1%和FE(丙醇)=4.8%进行计算。)
第3题 人工光合作用
人工光合作用研究旨在将太阳能储存在化学键中。光子通过激发敏化剂而被吸收,从而产生电荷分离态。激发的电子被转移到催化剂(析氢催化剂,HER)上,经两次还原后产生H2。常选的光敏剂或光吸收剂是[Ru(bpy)3]2+(bpy=2,2′-联吡啶),而HER通常采用钴配合物。
I:水分解过程的能量关系
3.1 计算反应H2(g)— 2H+(aq)+2e-的焓变。已知,质子的溶解焓ΔHaq(H+)=-1190 kJ mol-1;氢的电离能IE1=13.6 eV;H2的解离焓ΔHdiss(H2)=432 kJ mol-1。
理论上,25 °C时水电解成氧气和氢气的电压为1.23 V。由于此过程的TΔS值 >0,需要从环境吸收热量。如果提供超电压产生的热量能够补偿温度的降低,则该过程被称为热中和。已知25 °C时,H2O(l)的生成焓ΔH°H2O为-285 kJ mol-1。
3.2 计算25 °C时1摩尔H2O分解反应的熵变ΔS°R及达热中和时水分解反应的电压。
II:催化剂
钴-席夫碱(Cobalt-salen,salcomin,双水杨酰胺乙基钴,简称钴-席夫碱)型配合物是由质子和电子形成H2的潜在催化剂。钴-席夫碱的结构如图3-1所示:
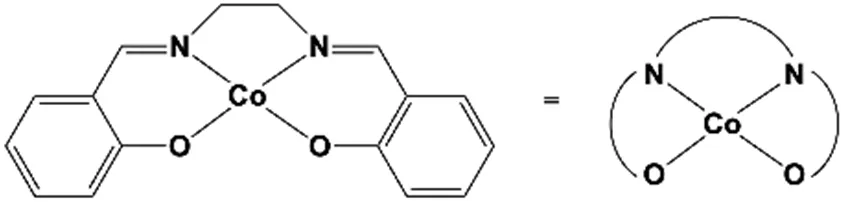
图3-1 钴-席夫碱的结构
3.3 确定钴-席夫碱中钴原子的氧化态以及钴中心周围的几何结构,从以下三种可能性中选择:四面体、平面四方形或八面体。
3.4 在溶液中,钴-席夫碱可与O2结合;O2通过与两个Co中心配位,将两个钴-席夫碱分子连接起来。因此,两个Co中心的氧化态就都是+III。画出产物结构。
H2的形成完全发生在钴中心。该反应可由一个4步催化循环描述,从Co2+开始,需要用到2个H+和2个电子。在其中的一个步骤中,通过分子内的电子转移形成氢化物。
3.5 画出催化循环的两种可能过程,并指出配合物的电荷和Co中心的氧化态(不应大于+III)。参照图3-2中的循环示例,用星号*标记氢化物形成步骤,并用字母C(化学反应)标记吸收H+的步骤,用字母E(电化学反应)标记吸收电子的步骤。[CoII]代表Cobalt-salen配合物。
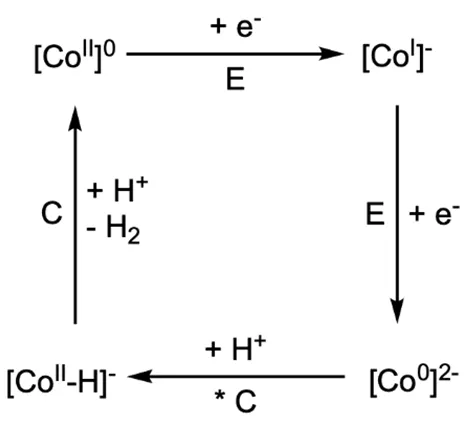
图3-2 3.5小题的循环示例
III:氧化还原电势
3.6 根据表3-1中给出的不同钴配合物的氧化还原电势,分别写出哪种配合物适用如下过程:

表3-1 氧化还原电对及其对应的标准电极电势([C2O4]2-=酸根,en=乙二胺)
a)中性pH下水的氧化;b)中性pH下水的还原。写出这两个过程对应的总反应(仅针对能够进行该反应的配合物),并计算中性pH下的电池电动势。在pH=7,T=298 K条件下,质子还原的半反应电极电势为-0.41 V。
IV:自然光合作用过程一瞥
生物H2的天然储存物是NADPH,它由NADP+结合氢负离子而成。NADPH的结构如图3-3所示。
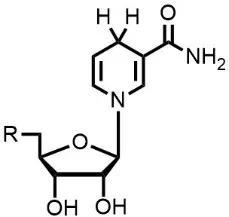
图3-3 NADPH的结构
3.7 画出NADP+的结构。
3.8 叶绿素在680 nm时的消光系数ε=8×104M-1cm-1。假设680 nm波长下,光子通量为100 nE s-1cm-2(1 E=6×1023光子)且光子到氢原子的传递效率为φ=20%。在1×1×1厘米的反应池中,要获得每秒1 nmol H2的转换频率,计算:a)每秒的光子数;b)所需要的叶绿素浓度。
第4题 含氟化合物和超价化合物
简介:氟基本上能与所有元素(包括惰性气体Kr和Xe)形成稳定且可分离出的化合物。含氟分子通常具有非寻常的结构。因此,氟与14–18族元素经常形成被称作超价的化合物。如今,含氟有机化合物的合成在很大程度上依赖于专门设计的试剂,如下的化合物4就是一个例子。
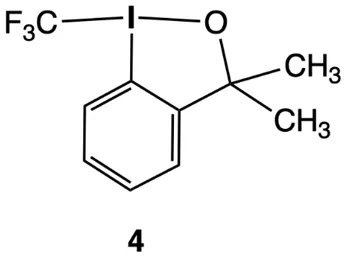
I 分子几何结构提示:E1–E8系列中的任何元素E很可能出现一次以上。
4.1 确定以下三个物种(1、[2]-和[3]-)中含有的元素E1、E2、E3和E4。第15–18族中部分元素的典型E-F键长范围列在表4-1中。
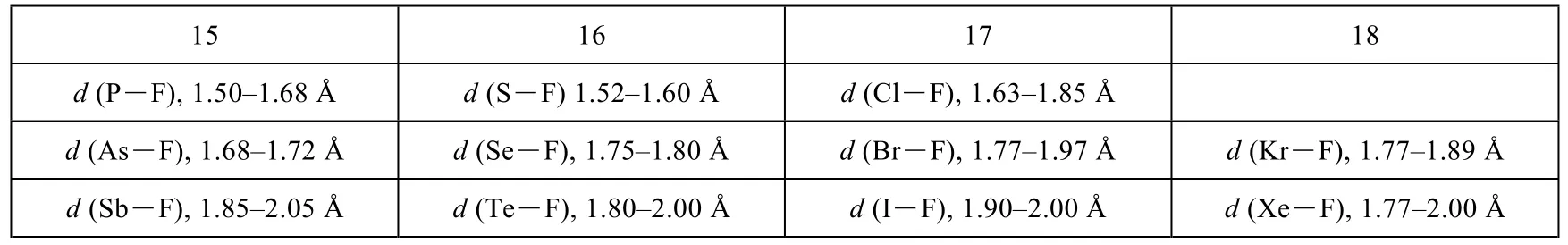
表4-1 第15–18族中部分元素的典型E―F键长范围
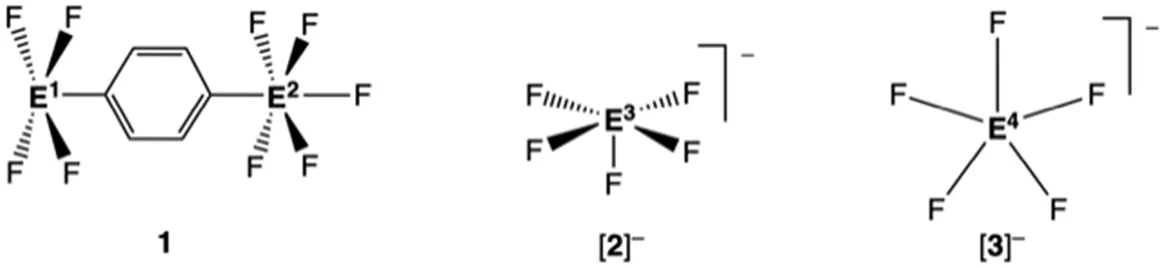
1:中性,非两性离子分子,E1,四方锥;E2,八面体,平均键长d(E1―F)=1.91 Å;平均键长d(E2―F)=1.58 Å[2]-:阴离子,四方锥,平均键长d(E3―F)=1.96 Å;[3]-:阴离子,平面五角形,平均键长d(E4―F)=1.98 Å
提示:
1.所给分子几何形状指的是与E1―E4成键的原子的排列方式。
2.分子1的元素分析表明其碳含量为17.75 wt.%。
4.2 若假设分子1是一个两性离子,在E1和E2处都带有1价的形式电荷,从而产生了假设的分子1'和1'',如下图所示。
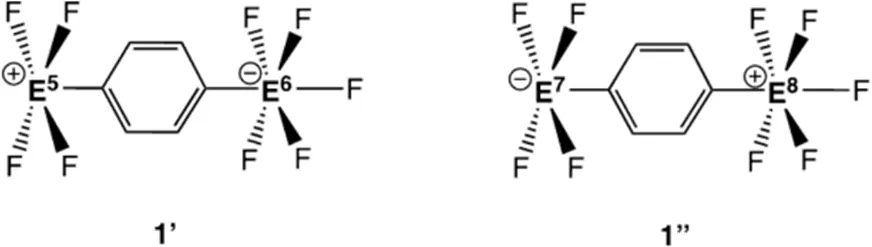
指出E5/E6和E7/E8分别代表哪些元素,才能使得分子1能满足题目中给出的几何形状,并且E―F键距离也与分子1的数据(参见表4-1)相近。
II 反应活性和结构
请看下图所示的反应:
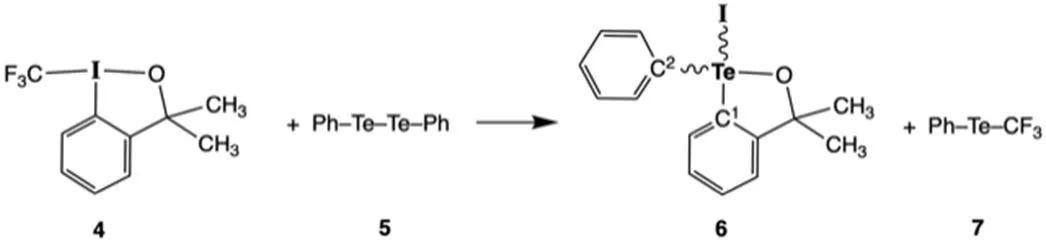
4.3 根据Te原子周围价层电子对的排列,判断化合物6的理想几何形状,从平面正方形、三角双锥、四面体、四方锥和八面体中做出选择。写出预期的理想键角C1―Te―I、C2―Te―I;I―Te―O和C1―Te―C2。
4.4 预测化合物4和6中两个甲基的1H NMR信号峰的数目。
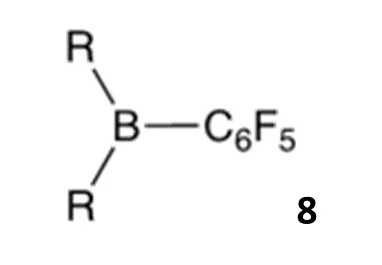
4.5 化合物6与AgF、(H3C)3SiCF3(简写为TMSCF3)依次发生反应。推断中间体A和最终产物B(二者均含碲)以及副产物C和D,画出A和B的几何结构,写出副产物C和D的化学式。(D的分子量为92.08)
假设化合物6与一种有较大空间位阻的、带有手性的、对映体纯的路易斯酸——例如已知的硼衍生物8——发生反应,如下图所示。该反应将生成一种新产物9,其组成相当于6和8的组合。进一步假定9是一种盐,其中阳离子源自6,阴离子源自8。
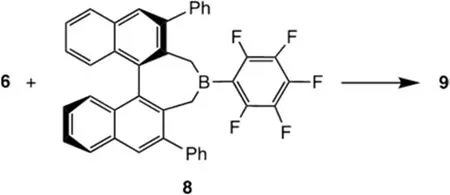
4.6 画出含碲的阳离子和含硼的阴离子的结构,并判断阳离子中价层电子对围绕碲原子分布的理想几何形状(在框中勾选)。提示:化合物8(手性,对映体纯)采用以下通用示意图表示:
□ 平面正方形
□ 平面三角形
□ 四面体
□ 三角锥
□ 三角双锥
4.7 写出盐类化合物9所有可能的立体异构体的数目。
III λ3-二氟碘烷的合成以及环绕单键的旋转
化合物12由起始原料10制得——在过量KF的存在下,10在干燥的乙腈中用三氯异氰尿酸(TCICA,11)进行氧化,制备方法如下所示:
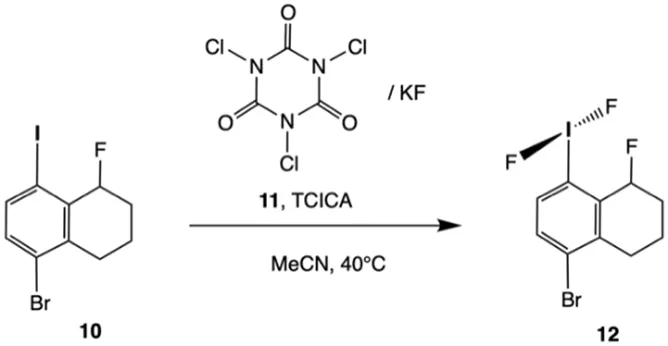
4.8 确定这一过程的电极半反应和电池总反应,写出反应方程式并配平。
提示:10缩写为R-I,12缩写为R-IF2,TCICA代表C3Cl3N3O3。TCICA的六元环在还原时保持完整。
4.9 化合物12中的IF2基团可以围绕I―C键旋转(可以想象一下分子螺旋桨,如下所示)。实验测得相应的旋转能垒为:Ea=30 kJ mol–1。又知,228 K下旋转的速率常数为k=2500 s–1。确定IF2基团在室温下(298 K)的旋转速率。将这一过程视为一个化学反应,确定其速率常数(单位为s-1)。

第5题 加氢脱硫
为降低污染环境的含硫化合物的排放,生产无硫燃料乃大势所趋。为了脱硫,炼厂采用氢辅助的加氢脱硫过程。
5.1 画出噻吩加氢脱硫过程(见图5-1)的产物A至E的结构。已知,A和B为环状的区域异构体,C是环状的。
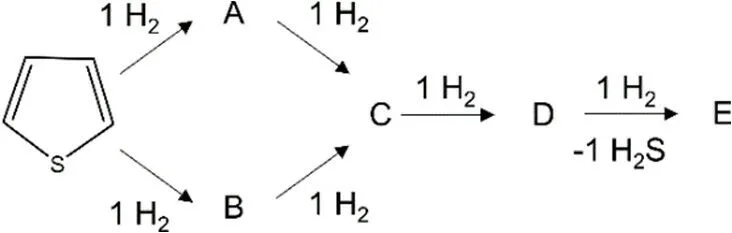
图5-1 噻吩加氢脱硫过程
硫有两种最常见的稳定天然同位素,32S和34S,二者的相对摩尔丰度分别是χ(32S)=94.8%和χ(34S)=4.37%。对氢而言,稳定的天然同位素是1H和2H(D),二者的相对摩尔丰度分别为χ(1H)=99.986%和χ(2H)=0.014%。
5.2 仅考虑上述同位素,写出H2S所有可能的同位素组成方式。
5.3 仅考虑上述同位素,写出H2S中同时含D和34S同位素的组成并计算其相对摩尔丰度,用百分数(%)表示。
脱硫是一种催化过程,通常在以SiO2为载体的MoS2(MoS2/SiO2)催化剂上进行。为了研究催化剂的表面,可以采用同位素交换方法。同位素交换反应发生在气固界面,只交换表面原子。在一级近似中,可认为体相原子不参与交换(图5-2)。
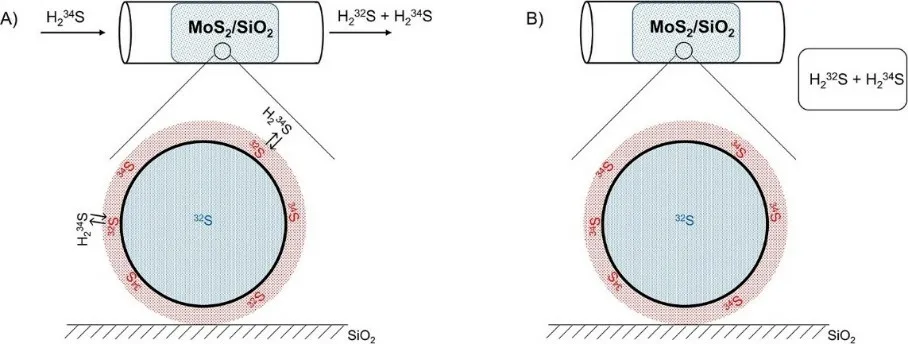
图5-2 实验过程(A)和最后阶段(B)的示意图
实验中,在流动床反应器中研究了MoS2/SiO2催化剂(初始只含32S,Mo质量分数wMo=4.280 wt.%)和同位素标记的气态H234S之间的同位素交换(图5-2)。将MoS2/SiO2催化剂(mcat=1.2350 g)置于Ar(作为平衡气)和H234S(H234S体积分数ϕH234S=1.00 vol.%,H234S同位素纯度α=99.95%)组成的混合气流(p=1.00 bar,v=20.0 mL min-1,T=23.0 °C)中。
实验持续时间为t=10.0分钟,在出口处收集整个实验过程产生的气体。在收集到的气体中,测得34S同位素占所有硫原子中的比例(γ)为γ=87.3 mol.%。假设气体均为理想气体,MoS2在表面上和体相中的元素(非同位素富集)组成相同,并且在实验结束时,处于表面的所有硫原子都与气相进行了交换。
5.4 计算交换的硫原子数n(S)surface(surface,表面)。以mol为单位。
如果你未能算出交换的硫原子总数,在以下所有计算中请使用数据1.53×10-5mol。
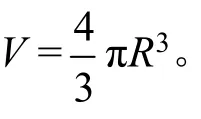
5.5 计算MoS2颗粒的半径R,以nm为单位。
实际上,同位素标记的原子会从表面扩散到体相,而未标记的原子则从体相扩散到表面,形成一个交换梯度(图5-3A)。因此,在给定的时间内,颗粒内部标记原子的比例会从粒子表面向中心递减。与此同时,随着交换时间的延长,体相原子参与交换反应的程度增加,如图5-3B所示。
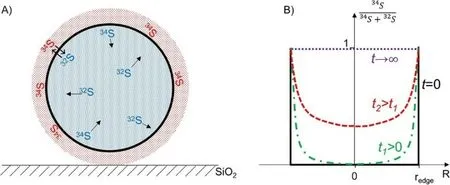
图5-3 A)MoS2颗粒中硫同位素从表面向体相扩散的示意图。表面的硫原子用红色表示,体相中的硫原子用蓝色表示。钼原子未显示;B)体相中34S原子的分数与时间和粒子中心距离的函数关系。redge对应于MoS2颗粒的半径
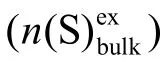
其中,t是交换实验的时间(如上所述),R是颗粒大小(球形颗粒的半径),D是扩散系数。基于电子显微镜对上述催化剂进行了研究,结果表明MoS2颗粒是均匀分布的球形,半径为35.0 nm。
5.6 取R=35.0 nm作为半径值并采用上述交换实验的数据,计算硫原子在MoS2中扩散的扩散系数D,答案以m2s-1为单位。在计算中,请使用以下近似值:当x<<1,ex≈ 1+x。
第6题 甲烷直接转化为甲醇
甲烷广泛存在于天然气中,是化学工业(如生产甲醇)中极具吸引力的原料。然而,由于甲醇比甲烷更容易被氧化,因此对这一过程的控制具有挑战性。
在化学循环过程中,载铜沸石催化剂的活性位点仅提供氧化成甲醇所需的单个氧原子并被消耗,从而避免了过氧化。在第二步中,催化剂在没有甲烷的情况下用氧气再生。下图给出了两种有潜力的铜催化位点(S1和S2)。在反应过程中,铜(II)被还原为铜(I)。
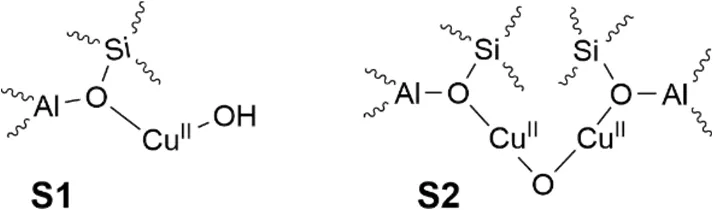
6.1 写出将一个甲烷分子氧化成甲醇时,所需S1和S2活性位点数。
在没有氧气的情况下,形成的甲醇不会从沸石中脱附。如果反应在恒温恒容的容器(如反应釜)中进行,压力下降仅来自甲烷的反应,甲烷可被视为理想气体。反应在1 L反应釜中进行,催化剂为200 mg担载4.3 wt.%铜的沸石,温度528 K,甲烷初始压力p0=933 Pa,反应完成后,压力下降到p∞=925 Pa。
6.2 计算发生反应的铜的百分比。
6.3 实验数据见图6-1。据此,可以判断CH4氧化反应(准)级数。在以下选项中选择正确说法。
□ 反应是(准)零级; □ 反应是(准)一级; □ 反应是(准)二级
6.4 写出在给定条件下与实验数据符合的CH4氧化的速率方程。注意,该方程可能与CH4的浓度、S1和S2活性位点浓度和反应速率常数相关。
6.5 在以下选项中勾选正确说法。
□ 至少有两种类型的铜位点发生反应,每一种都有不同的速率常数。
□ 在较高的温度下,载铜沸石氧化甲烷的速度更快。
□ 在较高的温度下,在反应完成后,铜表面上与甲烷反应的铜位点数量更多。
□ 其中一个反应在较高的温度下变慢。
电子顺磁共振谱(EPR)可观察到S1位点的顺磁性,而具有抗磁性位点的S2则不会产生EPR信号。EPR谱可测量电子自旋的数量。因此,S1活性位点的数量与EPR谱的二重积分I2成正比,即[S1]∝I2。在反应开始后的不同时间t,多个温度下T进行样品测量,获得电子顺磁共振谱图。
6.6 推导出关联I2(t)和kS1且对随时间呈现线性变化的方程,kS1是S1活性位点减少反应的速率常数。
6.7 在以下选项中勾选出需要用已知铜(II)标准物校准的测量。
□ 样品中顺磁性Cu(II)位点总数
□ 样品中顺磁性Cu(II)位点浓度
□ 速率常数
□ 样品中不同顺磁性Cu(II)物种的类型
通过EPR测量得知,528 K时S1活性位点反应的速率常数为2.604×10-3s-1。
6.8 结合图6-1,通过计算,确定甲烷与S2位点的反应是快于还是慢于其与S1位点的反应。
甲醇可通过不同的沸石催化剂进一步转化为有价值的烯烃。在此过程中,我们可以观察到摩尔质量为86.09 g mol-1的中间产物,元素分析结果为55.8 wt.% C,7.0 wt.% H,1H NMR谱由四个不同化学位移信号组成(a:12.2 ppm(1H,s),宽峰,加入D2O后消失;b:6.3 ppm(1H,d);c:5.7 ppm(1H,d);d:2.0 ppm(3H,s))。
6.9 画出中间产物的结构并标出a和d对应的氢。
美国能源部指定12种仅含C、H和O的化合物为基本化学品。这些化合物是最有前途的候选化合物,易于从可再生资源中制备,并且可以从中制备出多种目标衍生物。其中之一是化合物A,它可以进一步衍生或用于医药或洗涤剂等。
A的摩尔质量为156.03 g mol-1。元素分析(EA):46.15 wt.% C,2.56 wt.% H。核磁分析结果如下:在DMSO中1H NMR:7.81 ppm(a,s),13.0 ppm(b,s,宽峰,当加入D2O时消失),两个信号积分相同;13C NMR:165.1 ppm(1),150.6 ppm(2)和120.6 ppm(3)。
6.10 给出A的一种可能结构式,并根据化学位移指认其中所有H(标a和b)和C1。
第7题 酶动力学
米恰利-门顿(Michaelis-Menten,MM)机理于1913年提出,用于描述酶催化的动力学。在该机制中,酶E催化底物S转化为产物P。
遵循米氏机理的酶促反应的初始速率通常为:
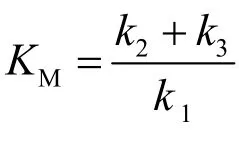
注意:第7.1和7.2小题可以有一个、多个或没有正确答案。
7.1 对于方程式(1)和(2)中所表示的初始反应速度v0,在以下选项中勾选出正确的等价表示方式,[ES]max代表络合物ES的最大浓度。
7.2 在以下选项中勾选预计其(yvs.x,即y随x的变化)呈现线性关系的参数对(一对或多对)。
许多酶催化反应是多底物而非单底物转化。然而,如果其中一种底物的浓度远高于另一种底物的浓度或保持不变,米氏机理也是有效的。下面我们将研究遵循该机理的两个独立的酶反应体系。
酶反应1
底物A和B在酶E的催化下生成产物PA与PB。当游离酶和所有酶-底物配合物之间快速达到平衡时,v0可用下式描述:
k是其中一个基元反应的速率常数。相同的平衡常数K表达了两种底物具有相同的从E的相应活性位点解离的特性。
7.3 如果底物B浓度保持恒定为c0,请将上式(3)转变成米氏方程常用形式(1)中的形式,并给出vmax的表达式。
7.4 对于酶反应体系1,导出其符合式(3)的动力学机理,要体现所有中间体和产物。指出速率常数k对应的反应。
酶反应2
酶E有五个活性位点,每个位点对底物SA、SB、SC中的一种有特异性,分别选择性地转化为产物PA、PB或PC。每种底物至少对应于一种活性位点。每个活性位点都相互独立。
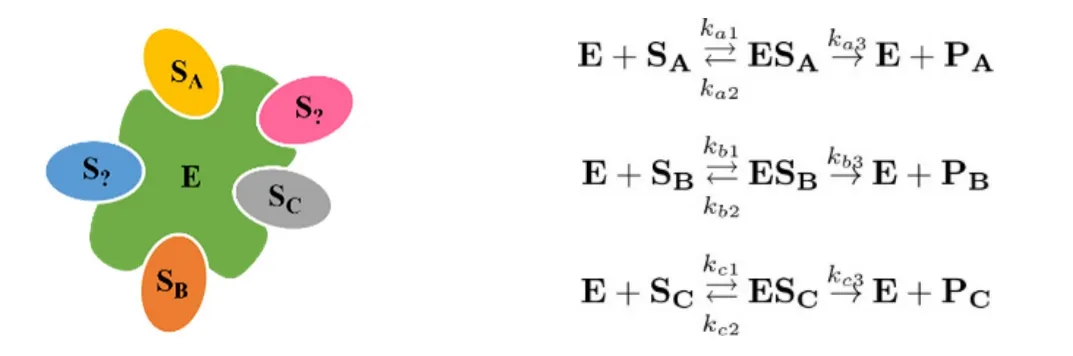
对于E,已知:
1.与底物SC的结合能力高于SB。
2.v0i与v0i/[Si]0的关系称作Eadie-Hofstee曲线,如下图所示(省略了坐标尺度),此处v0i与v0i/[Si]0分别表示不同底物SA、SB、SC的初始反应速率(v0i)和单位活性位点的初始反应速率(v0i/[Si]0)。
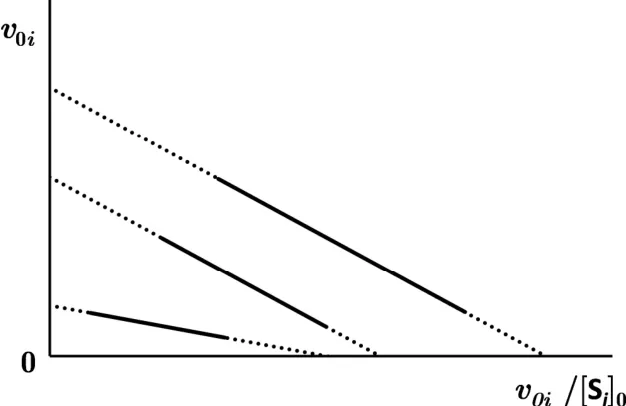
3.当E的活性位点被底物SA、SB、SC饱和后,对于SC,每个活性位点催化转化数为10200 min-1,每秒合成的PA、PB、PC分子数共2023个。同时,每小时检测到的PA和PB分子总量不超过5.94×106个。
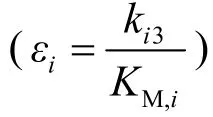
5.两个配合物ESi解离成E和Si的速率常数相等。ESC分解成起始物的活化能比生成最终产物的活化能高1266 J mol-1,设两个反应的指前因子相等,T=25 °C。
6.对于E+Si的反应:kc1=1.57×107M-1s-1,ka1=kb1。
7.5 填写如下表格,并给出计算过程。提示:
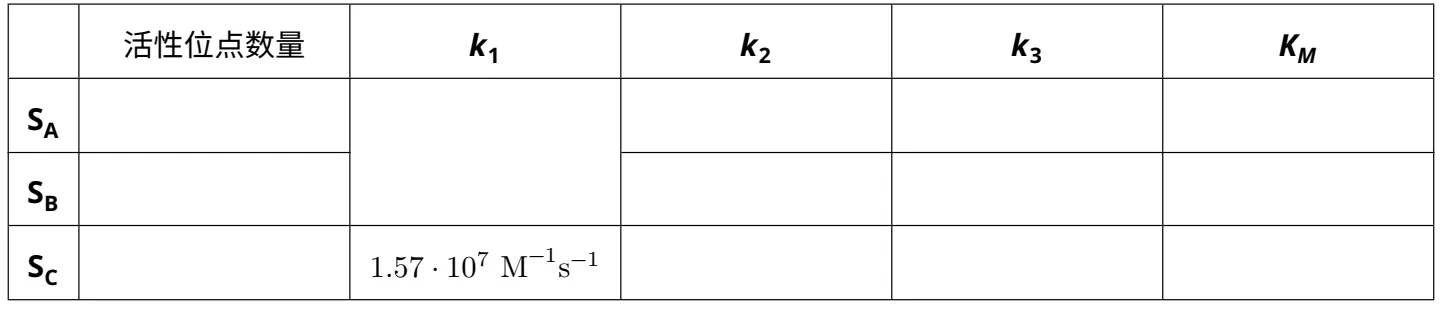
?
基于1.和2.的信息,确定KM,A、KM,B和KM,C(<,>,=)的大小关系。
基于3.和5.中的信息,可以填写完成表格第一列(每种底物的活性位点数)和最后一行(底物SC的所有常数)。确认活性位点数的总和等于5。
第8题 Nazarov反应
Nazarov反应是由二烯基酮转化为环戊酮的一类常见反应。它是在光照下或酸催化下的电环化反应。反应先形成五元环,接着再质子转移形成产物。
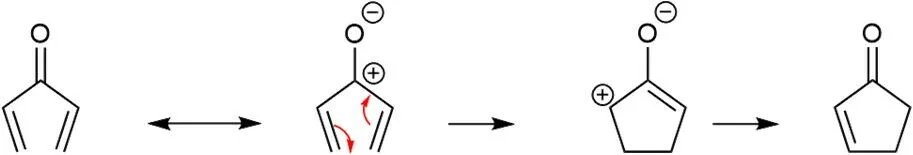
8.1 画出Nazarov反应底物π电子的前线分子轨道,在相应的轨道上填入电子。在合适的方框中用X标记:i)HOMO(最高占据分子轨道)和 ii)LUMO(最低未占据分子轨道)。对于本小题,可以将二乙烯基酮等同于戊二烯基阳离子。
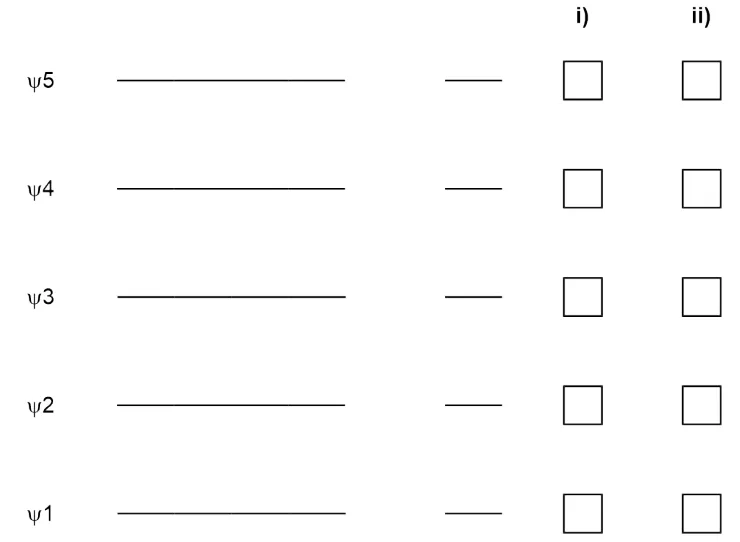
8.2 根据题8.1中你所画的前线分子轨道,判断以二乙烯基酮为底物的Nazarov反应,与加热条件下对应的是顺旋还是对旋;与光照条件相对应的是顺旋还是对旋。在下表中,用X在框中标记与反应条件相对应的轨道旋转方式。

?
8.3 Nazarov反应是合成天然产物Farnesin的关键反应。依据以下两个反应条件,为产物A和B分别画出一种可能的立体结构式(要求立体化学)。请注意,所有可能产物的1H NMR图谱中在化学位移6.70–6.73 ppm处均有一个信号峰。

如下图所示,Capnellene的合成以不饱和醛C为原料。原料C在条件D下反应,接着再与负载在碳上的MnO2反应,生成如下图所示的二乙烯基酮E。E在P2O5和甲磺酸MsOH的溶液中反应形成F。F再经过三步反应转化为不饱和酮I。
8.4 在下列选项中选择哪些可用于将C转化为E的反应中(反应条件D)。
□ H2C=CHMgBr
□ 1.NaBH42.H2C=CHLi
□ H2C=CHBr,Pd(PPh3)4
□ H2C=CHMgBr,CuI
8.5 画出中间产物F、G和H的立体结构式(要求立体化学)。
然后,将烯酮I在THF溶液中与H2C=CHMgBr和CuI反应,形成中间产物J,随后再进行臭氧化反应,形成中间产物K。K在1H NMR图谱中化学位移9.61 ppm处有一个信号峰。K与5% KOH在THF和乙醚溶液中反应,形成中间产物L。在H2气氛下,利用Pt催化剂催化氢化,形成M,最终转化为Capnellene。
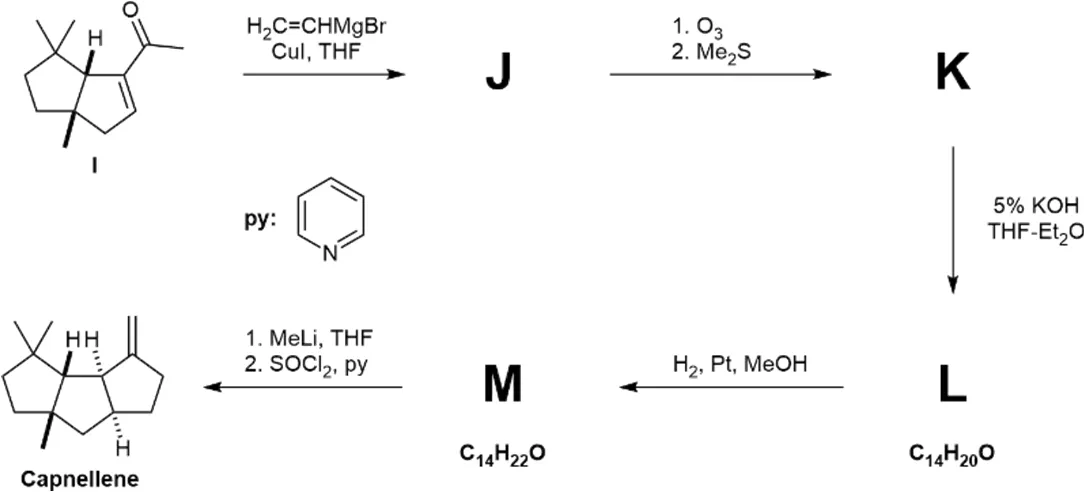
8.6 画出J、K、L和M的立体结构式(要求立体化学)。
第9题 有机合成中的电化学合成
Kolbe电解反应是指两种羧酸在电解下的脱羧二聚反应,而且只能在羧酸去质子化后才能反应。未配平方程如下式所示:
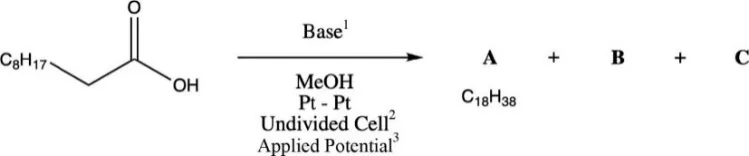
1:碱;2:一个电解槽;3:电极电势
反应过程中会产生两种气体(B和C)。气体B可与Ca(OH)2发生反应,而气体C易燃。
9.1 画出A、B、C的结构式。
9.2 此合成实际上是氧化还原反应,其中羧酸被氧化。画出氧化和还原半反应以及完整氧化还原反应的方程式。
9.3 在方框中画出氧化脱羧形成产物的过程中中间体和产物的结构式。
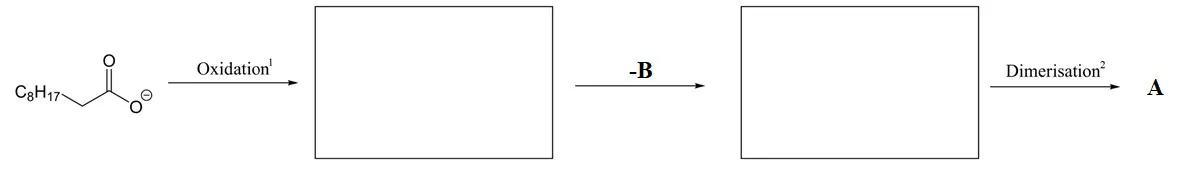
Kolbe电解反应通常只能使用长链脂肪羧酸作为底物,而不用使用某些如D等特殊的羧酸。这是由于在反应过程中自由基中间体E会被过度氧化为正离子F。
中间体F可与亲核试剂反应形成各种副产物。例如,与D反应形成酯G,与MeOH反应形成H。
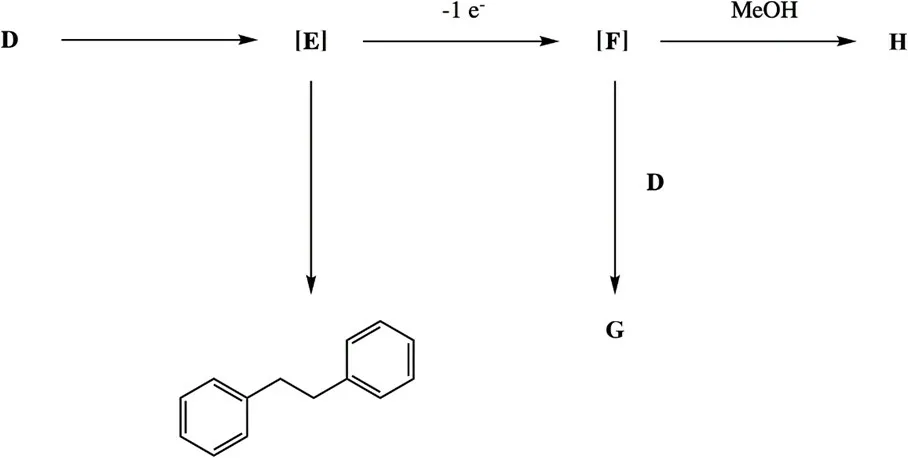
9.4 画出D–H的结构式。
在过量乙酸J存在下,羧酸I的电解反应会形成两种在硅胶上无法分离的主要产物(通过1H NMR分析)。它们的1H NMR图谱几乎一致。在1H NMR谱中,这两种物质只能通过化学位移微小的差别进行区分。这两种产物的1:1混合物的1H NMR谱如下:
1H NMR(K和L):4.18–4.08(m,4H),3.95–3.60(m,6H),3.43(dt,2H,J=7.8,2.2 Hz),2.55–2.25(m,4H),2.20–1.95(m,2H),1.65–1.50(m,2H)。
化合物K的特征图谱:1.26(t,3H,J=7.2 Hz),1.20(d,3H,J=6.6 Hz)。
化合物L的特征图谱:1.24(t,3H,J=7.2 Hz),1.15(d,3H,J=6.6 Hz)。
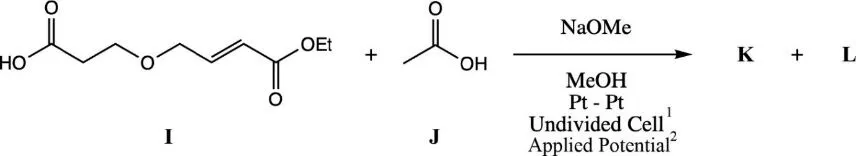
1:一个电解槽;2:电极电势
9.5 画出产物K和L的结构式。判断这两个产物的立体化学关系。
电极材料会影响有机电化学合成反应的选择性。与电极表面的强作用力有利于分子间反应。苯甲醛(M)(16 mM,在1 M KOH水溶液中,阳极为Pt,电极电势相对于Ag/AgCl为-1.3 V)的还原电解反应,会由于所使用的不同阴极材料而形成不同的产物。下图显示了不同阴极材料下的两种产物比例以及它们的质谱图。
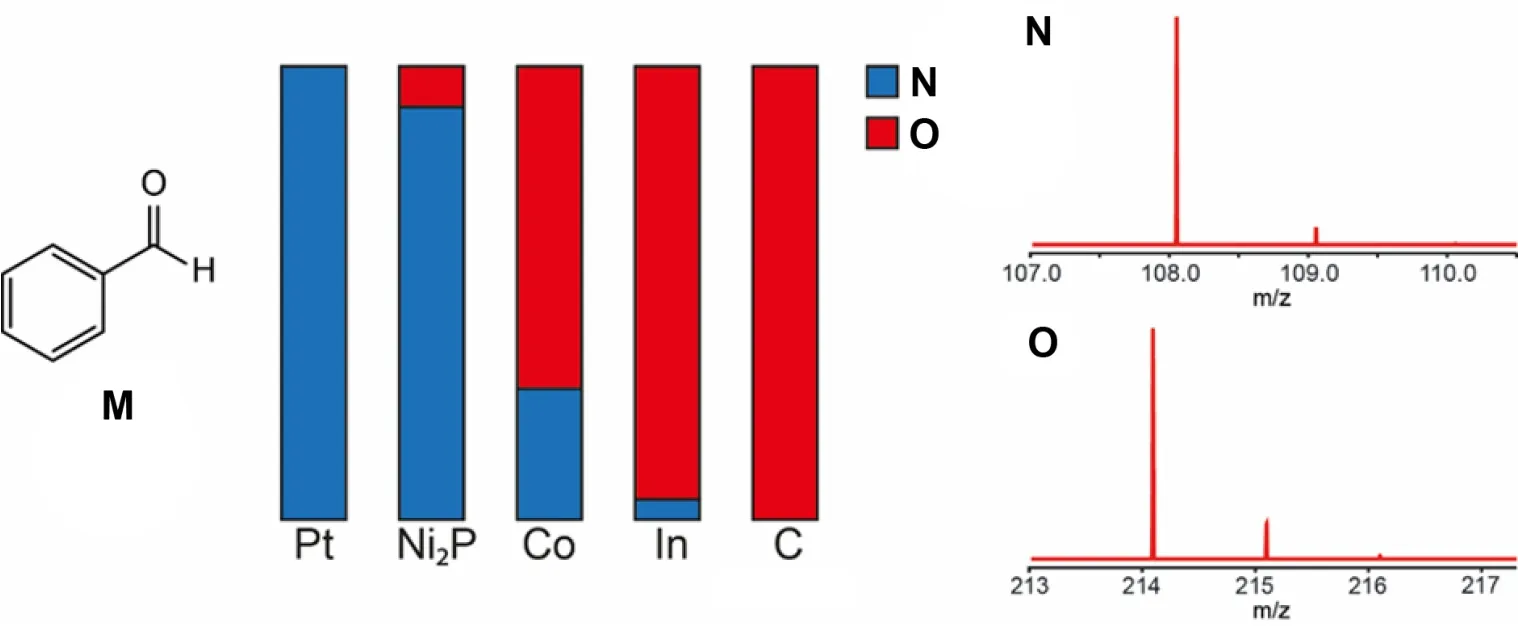
9.6 画出产物N和O的结构式。
烯烃,如烯醇醚等,也可以氧化偶联。这通常包含了以下基元反应:烯烃碳碳双键的阳极氧化产生自由基正离子,接着自由基正离子与亲核试剂反应。
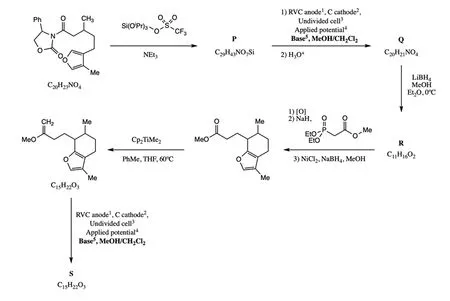
1:RVC=玻碳阳极;2:碳为阴极;3:一个电解槽;4:电极电势;5:碱cc
9.7 画出化合物P、Q、R和S的结构式,不要求立体化学。提示:S为三环产物。
第10题 瑞士——制药之国
Pasireotide(1)是瑞士诺华制药公司开发的一种多肽类药物,用于治疗库欣病。

10.1 判断Pasireotide(1)一共有多少个手性中心,并计算该分子一共有多少个立体异构体。
Pasireotide(1)是一种环肽。如合成路线图10-1所示,其合成的关键中间体(线性多肽2)可以使用Fmoc/tBu策略通过固相多肽合成法(SPPS)合成。
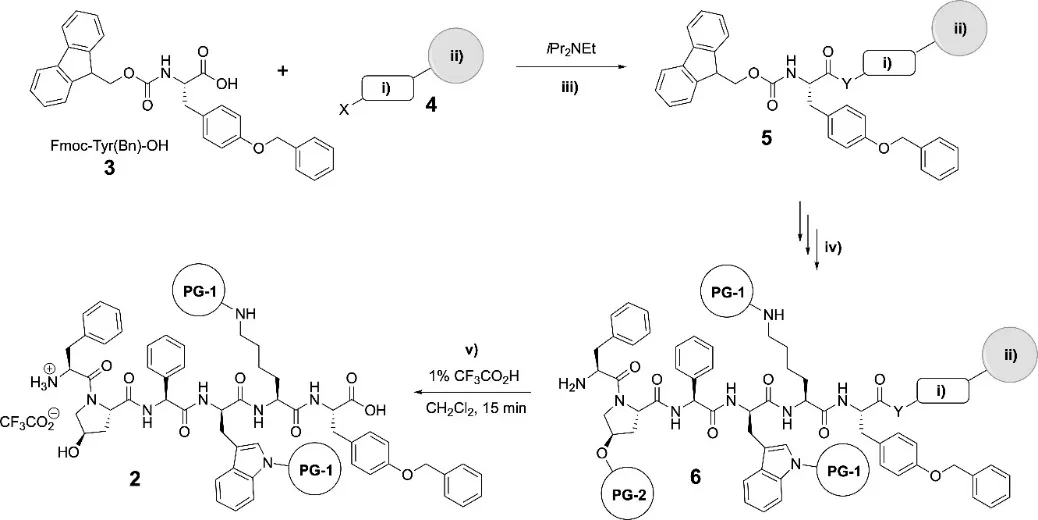
图10-1 肽2的固相合成路线
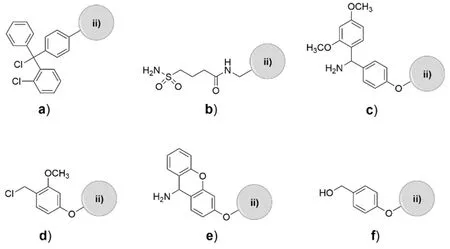
图10-2 树脂4的结构 ii)树脂 a)邻氯苯基-二苯基-氯甲基树脂;b)安全锁树脂;c)Rink 酰胺树脂;d)SASRIN -氯树脂;e)Sieber酰胺树脂;f)王氏树脂
以Boc-Tyr-OH(7)为起始原料合成Fmoc-Tyr(Bn)-OH(3)的路线如下:

10.2 画出上图所示的Fmoc-Tyr(Bn)-OH(3)合成路线所使用试剂A和D以及中间体B和C的结构式。
在中间体2的固相合成路线中,首先需要将Fmoc-Tyr(Bn)-OH(3)与带连接片段的树脂相连接。
10.3 根据题目中合成路线图10-1所提供的信息,判断上述六种树脂中哪些可用于肽2的固相合成。在以下选项中选择正确答案。
□ 2-氯三苯甲基氯树脂(a)
□ 安全锁树脂(b)
□ Rink酰胺树脂(c)
□ SASRIN-氯树脂(d)
□ Sieber酰胺树脂(e)
□ 王氏树脂(f)

10.4 上图列出了6个保护基g–l。根据试卷中图10-1的合成路线所提供的信息,在Pasireotide(1)中的其他保护基能正常保留的情况下,最适合作为2的固相合成过程中侧链保护基PG-1和PG-2分别是上述六个中的哪一个?分别只有一个答案是正确的。
接下来,如图10-3所示,线性肽2经分子内偶联反应形成环肽8:
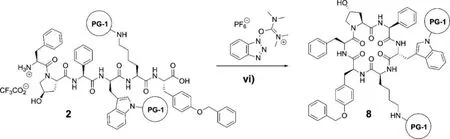
图10-3 环肽8的合成路线;vi)碱
10.5 有关肽2环化形成8的下列表述中,哪些是正确的?在以下选项中选择正确答案。相关结构如图10-4所示。不正确的答案会被扣分,但总分不能为负。
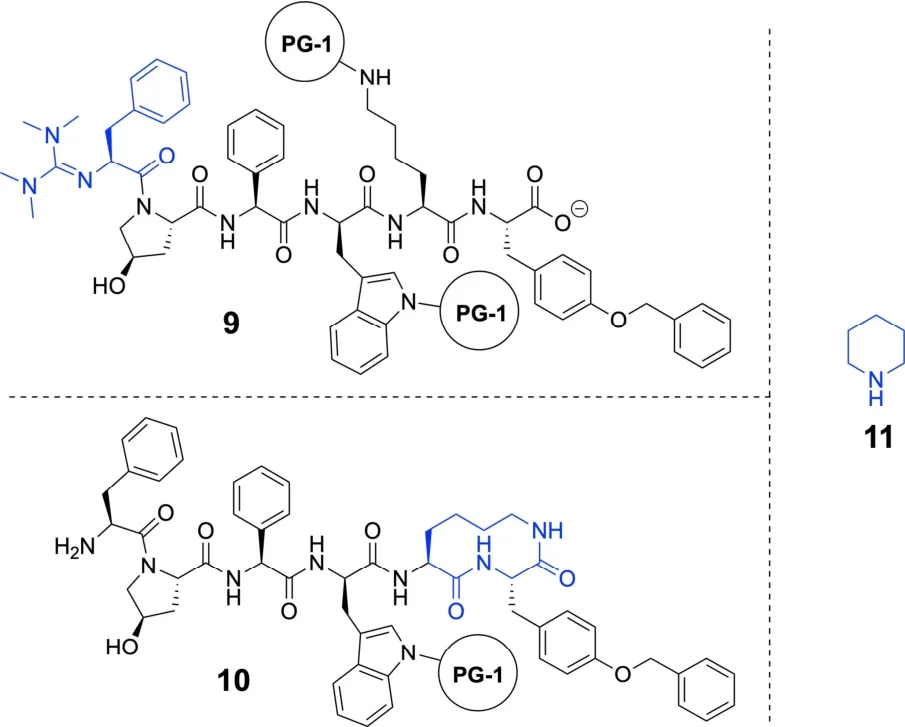
图10-4 题10.5的相关结构
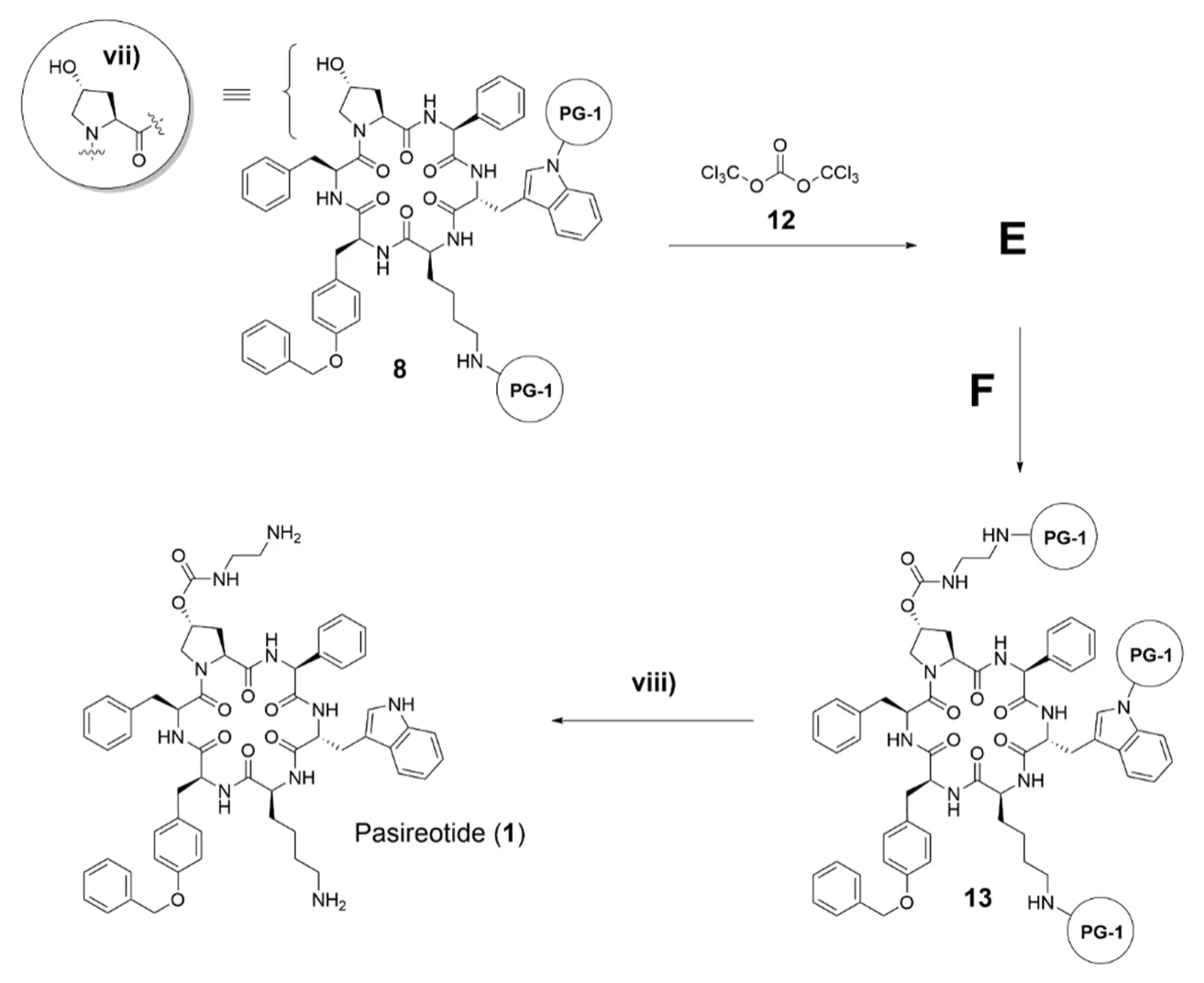
图10-5 vii)可以将8缩写为圆圈中的结构式;viii)脱除保护基团
□ 该反应一个可能的副产物是苯丙氨酸残基N末端四甲基胍基化,生成化合物9。
□ 该反应一个可能的副产物是保护基PG-1的裂解,并通过赖氨酸残基的氨基环化,形成化合物10。
□ 反应必须在底物肽的高浓度下进行才能达到足够高的反应速率。
□ 反应必须在底物肽的低浓度下进行,以防止形成多聚体。
□ 哌啶(11)在该反应中合适作碱。
合成路线中的最后步骤包括了肽8中4-羟基脯氨酸片段中OH基团的官能化,然后脱除所有保护基团,得到Pasireotide(1)。
10.6 画出中间体E的立体结构式(要求立体化学)和试剂F的结构式。在画E的结构式时,可以用圆圈中的结构简式代表中间产物8,在画F的结构式时,可以直接使用合成路线5中所示的PG-1保护基团。
10.7 确定将8完全转化为13所需要的化合物12的最少当量数为多少。
补充材料:考试规则、常数和谱图指认信息以及题目赋分与占比见补充材料,可通过链接https://www.dxhx.pku.edu.cn 免费下载。
猜你喜欢
云南化工(2021年6期)2021-12-21
上海金属(2021年6期)2021-12-02
昆明医科大学学报(2021年3期)2021-07-22
科学(2020年2期)2020-08-24
天然产物研究与开发(2019年10期)2019-11-05
生物学通报(2019年3期)2019-02-17
中国民族医药杂志(2016年2期)2016-05-14
生物技术通报(2015年1期)2015-04-10
赤峰学院学报·自然科学版(2013年4期)2013-07-31
天津医科大学学报(2011年4期)2011-07-13
