
上市小分子抗病毒药物研究进展
2024-01-23 01:30薛圣志毕金龙惠新平武全香
大学化学 2023年12期
薛圣志,毕金龙,惠新平,*,武全香,*
1 兰州大学化学化工学院,兰州 730000
2 兰州大学生命科学学院,兰州 730000
病毒是一类严重危害人类生命健康的病原体,具有传播速度快、传播力强等特点。目前,新型冠状病毒感染(COVID-19)正在全球流行,被世界卫生组织列为紧急情况下优先研究的疾病。虽然抗病毒药物研发取得了较大进展,但现有的抗病毒药物和疗法仍有较多局限,无法满足临床应用需求,研发新机制和新结构的抗病毒药物迫在眉睫。
抗病毒药物按照分子量不同分为小分子药物和大分子药物。小分子药物以给药方式灵活,可口服;潜在作用靶点较多,机制多样,可进入细胞膜;生产成本相对较低,质量控制相对较好等优势占据研发领域的绝大部分。近期更多的新模式和新生物方法为小分子药物的发现和研究带来了便利,大多数制药公司都在以更多样的方式进行药物研究[1]。目前,每年由美国食品药品管理局(Food and drug administration,FDA)批准的抗病毒药物有3–4种,其中小分子药物2–3种[2]。新型冠状病毒感染和艾滋病是人类重点关注的疾病,病毒引起的流行性感冒作为一种全球重点公共卫生问题,传播广泛,易爆发流行,抗病毒药物研究受到了人们的广泛关注。本文介绍了2020年以来上市的抗新型冠状病毒和抗艾滋病毒药物的研究进展,同时介绍了一种最近由FDA批准的抗流感病毒药物。
1 抗新型冠状病毒药物
新型冠状病毒是一类具有包膜、基因组为线性单股正链的核糖核酸病毒,外膜存在多种跨膜蛋白,通过结合宿主细胞受体感染。病毒进入细胞后直接以自身遗传物质为模板指导蛋白质合成,并通过RNA聚合酶生成负链模板,进行基因组的扩增和病毒结构蛋白的合成,装配完成后通过胞吐的方式完成复制。抗新型冠状病毒药物研究的作用靶点主要集中在病毒包膜表面刺突蛋白的中和抗体和病毒复制周期关键酶抑制剂,药物通过阻断刺突蛋白与受体结合或影响病毒复制过程实现抗病毒。目前,抗新型冠状病毒药物,如莫努匹韦和瑞德西韦都是核苷类似物,作用人群有明显局限,较多药物相互作用更限制了其应用。FDA批准或授权的莫努匹韦和瑞德西韦可抑制病毒复制周期,帕克斯洛维德作为代表性口服药物在新型冠状病毒感染的临床治疗中取得了较好效果。
1.1 莫努匹韦
莫努匹韦(Molnupiravir,商品名Lagevrio,1,图1)是由美国默克(Merck)公司研发,2021年11月4日由英国药品和保健品管理局批准,全球第一个上市的治疗新型冠状病毒感染药物,主要用于治疗具有发展为中症和重症新型冠状病毒感染风险的轻、中度成人患者。

图1 莫努匹韦、EIDD-1931和EIDD体内三磷酸化产物的结构
1.1.1 前药修饰过程
莫努匹韦是β-D-N4-羟基胞苷(EIDD-1931,2,图1)的前药[3]。研究发现,EIDD-1931虽具有良好的抗新型冠状病毒活性,但其结构中存在多个羟基和氮原子,亲水性较强,体内代谢迅速。为此,研究者将核糖5’-碳上的羟基用异丁酸酐酯化,降低了亲水性,进而降低体内代谢速度,延长药物作用时间,得到了临床治疗效果良好的莫努匹韦。
1.1.2 药物作用机制
如图2所示,莫努匹韦在人体内转化为核苷酸类似物2(图1),三磷酸化产物3(图1)在细胞内参与病毒的RNA转录过程。3作为三磷酸化核苷酸类似物转录完成的病毒RNA存在突变,不能表达出正常蛋白质,影响病毒复制周期,使体内病毒载量保持在低水平,实现抗病毒作用[4,5]。
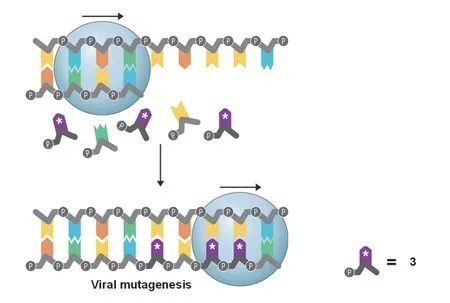
图2 莫努匹韦体内系列转化后参与病毒RNA合成过程
1.1.3 莫努匹韦的合成
莫努匹韦的合成以尿嘧啶核苷(4)为原料(图3),首先在酸性条件下用丙酮保护五碳糖的邻二羟基,同时氨基成盐。然后用异丁酸酐使5’-位酯化和4-位酰胺化,之后在羟胺溶液中得到化合物7,最后脱保护得到莫努匹韦[6]。分析发现,莫努匹韦以及体内活性物质β-D-N4-羟基胞苷比尿嘧啶更易与腺嘌呤形成氢键,通过竞争配对,以代谢拮抗方式实现抗病毒。

图3 莫努匹韦的一种合成路线
1.2 帕克斯洛维德
帕克斯洛维德(Paxlovid)是由美国辉瑞制药公司(Pfizer)研发,2021年11月22日FDA批准获得紧急使用授权。它主要含有两种药物:奈玛特韦(Nirmatrelvir,8,图4)和利托那韦(Ritonavir,9,图4)。此药用于治疗SARS-CoV-2病毒检测阳性且症状为轻至中度的成人和儿童,患者必须≥12岁、体重≥40 kg,是目前最为广泛使用的治疗药物。奈玛特韦是SARS-CoV-2的3CL特异性蛋白酶(3CL Protease,3CLpro)抑制剂,而利托那韦是蛋白酶抑制药,对于SARS-CoV-2的3CLpro没有活性,小剂量利托那韦有助于减缓奈玛特韦代谢或分解,增加其血浆浓度。
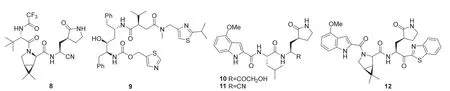
图4 奈玛特韦、利托那韦及奈玛特韦结构修饰关键中间体的分子结构
1.2.1 药物发现和奈玛特韦的修饰
2003年,在SARS-CoV-1流行期间,Hoffman等人[7]开发了对3CLpro具有强抑制作用的羟甲基酮衍生物(PF-00835231,10,图4)。因SARS-CoV-2和SARS-CoV-1的3CLpro序列的活性位点具有96%和100%同一性,他们研究用该化合物治疗COVID-19。化合物10活性好且代谢稳定,但渗透性和口服利用度低,难以口服给药。由于较多的氢键给体与生物利用度低有关,他们将10的α-羟甲基酮修饰为氰基得到化合物11。11的口服利用度提高,但活性明显降低。随后,引入苯并噻唑和环丙烷并四氢吡咯结构得到化合物12。12的渗透性显著提升,但因环丙烷并四氢吡咯结构单元占据了疏水口袋,导致活性远不如10。研究发现12的吲哚结构单元并没有很好地占据口袋,进而将吲哚修饰为甲基磺酰胺,进一步优化为三氟乙酰胺,并将苯并噻唑修饰为氰基得到了奈玛特韦[8]。
1.2.2 作用机制
3CLpro是SARS-CoV-2在生物体内复制的一种关键蛋白酶,抑制3CLpro的活性是抑制SARSCoV-2复制的理想手段。如图5所示,奈玛特韦进入细胞后,与3CLpro共价结合,并占据其切割长肽链的结合位点[9],从而抑制3Clpro活性,进而抑制pp1a和pp1ab蛋白水解,阻止病毒复制。
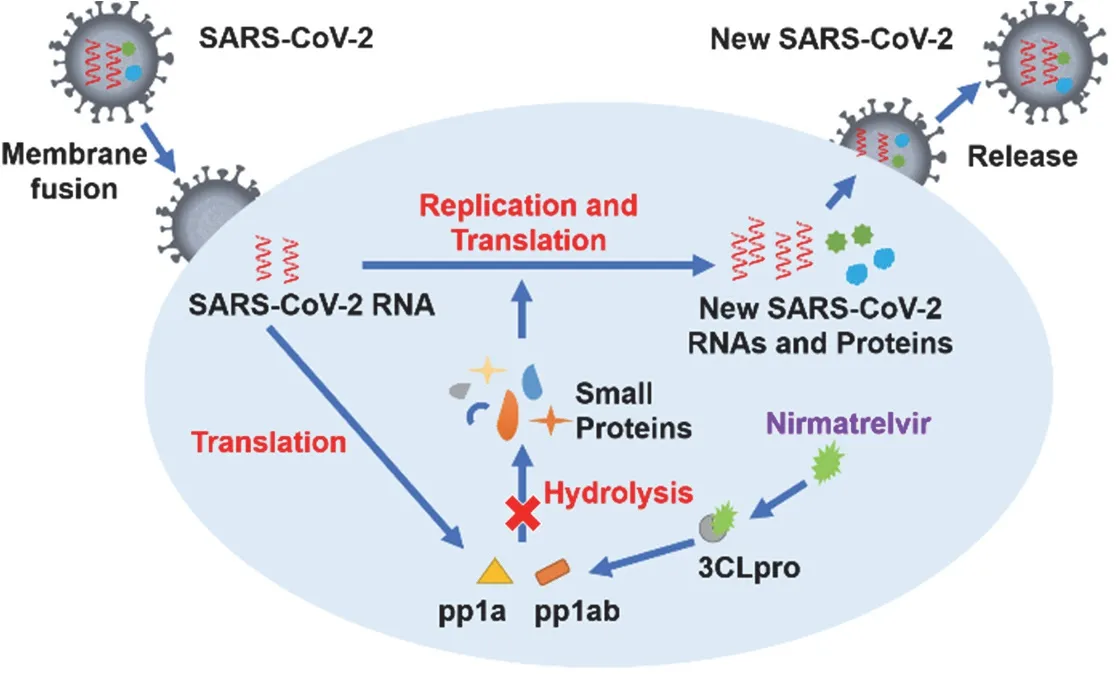
图5 奈玛特韦的作用机制
1.2.3 构效关系
在结构上,SARS-CoV-2的3CLpro有严格的谷氨酰胺底物偏好。奈玛特韦分子中,吡咯烷酮作为谷氨酰胺侧链的模拟物。吡咯烷酮在谷氨酰胺侧链的较高柔韧性上提供刚性,作用时通过减少结构变化以降低构象熵损失,提高反应活性。在结构修饰中,6,6-二甲基-3-氮杂双环[3.1.0]己烷结构阻碍了氢键形成,提高了药物口服利用度。相对惰性的氰基提高了与3CLpro蛋白结合时的选择性[10]。
1.2.4 奈玛特韦的合成
Owen等人[11]报道了奈玛特韦(8)的合成。如图6所示,(1R,2S,5S)-6,6-二甲基-3-氮杂双环[3.1.0]己烷-2-羧酸酯(15)发生酰胺化反应得到16,经过水解、脱保护得到17,氨基三氟乙酰化得到18,18与化合物14形成酰胺键,最后使用伯吉斯试剂使酰胺脱水生成氰基,得到奈玛特韦。
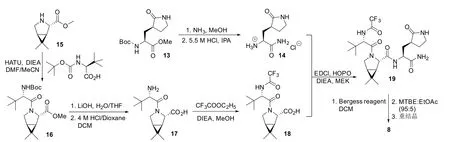
图6 奈玛特韦的合成路线
1.3 瑞德西韦
瑞德西韦(Remdesivir,商品名:Veklury,20,图7)是由美国吉利德科学公司(Gilead)研发,2020年10月22日FDA批准上市,用于治疗新型冠状病毒病住院成人和儿童患者。
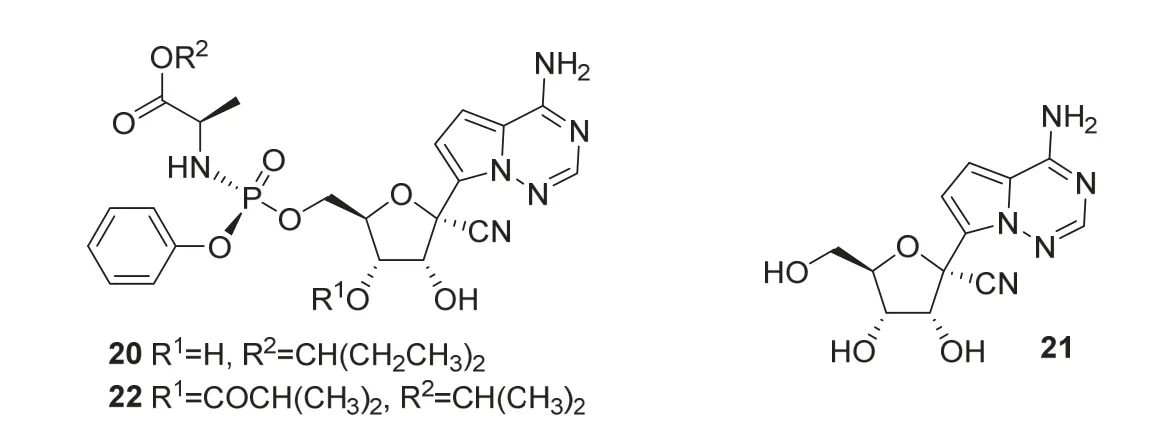
图7 瑞德西韦和药物修饰中间体的结构
1.3.1 药物修饰过程
瑞德西韦由修饰核苷类药物GS-441524(21,图7)得到,是一种具有广谱抗病毒活性的1’-氰基取代C-核苷核糖类似物[12],C-核苷更能耐受人体代谢。但大多数核苷膜透过性低,借鉴药物索菲布韦,将5’-位羟基修饰为氨基膦酰酯得到GS-6620(22)。膦酸化提高了药物在人体内的转化率,同时氨基膦酰酯有助于药物穿过细胞膜进入细胞,释放核苷单磷酸[13,14]。通过对RNA病毒药物的高通量筛选,得到了前药瑞德西韦。
1.3.2 作用机制
瑞德西韦和莫努匹韦均为核苷类药物,二者均可干扰病毒DNA到RNA的转录过程,瑞德西韦在体内的活性形式可与病毒的RNA依赖性RNA聚合酶(RNA-dependent RNA polymerase,RdRp)结合。如图8所示,由于核糖5’-位羟基膦酸化,从而实现了转录过程的提前终止,导致RNA结构破坏[15],影响病毒复制,从而降低病毒浓度,达到抗病毒效果。
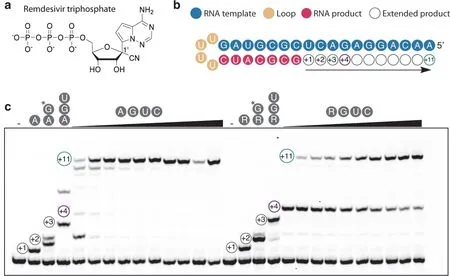
图8 瑞德西韦的作用机制及验证
1.3.3 药物构效关系
构效关系研究表明,瑞德西韦中核糖5’-位的氨基膦酰胺侧链可以提高细胞渗透率,单膦酸酯可以提高三膦酸化的速率。1’-位氰基可以提高对病毒聚合酶抑制的选择性,1’-位取代基的极性和位阻影响药物毒性。碱基选用非天然胞嘧啶C-核苷降低了体内代谢损耗,其他碱基会降低活性[14]。
1.3.4 瑞德西韦的合成
如图9所示,氧化半缩醛23得到24,7-碘吡咯并[2,1-f]三嗪-4-胺(25)在正丁基锂和三甲基氯硅烷作用下与24发生加成反应得到26,26与三甲基氰硅烷发生取代反应形成27,随后脱苄基、手性拆分得到28,最后用氨基磷酸氯29酯化得到瑞德西韦[16]。
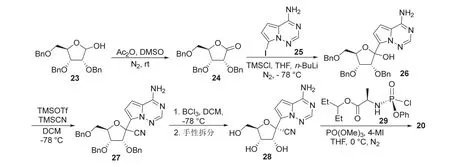
图9 一种合成瑞德西韦的路线
1.3.5 抗新型冠状病毒药物活性比较
新型冠状疫情爆发初期,发现多种药物具有抗病毒活性。法匹拉韦作为广谱抗RNA病毒药物对新型冠状病毒也具有活性,具有抗疟活性的磷酸氯喹也表现出抗新型冠状病毒活性而入选初期治疗药物。如表1所示,与疫情初期使用的磷酸氯喹和法匹拉韦相比,瑞德西韦具有高的半数有效浓度(Half-effective concentration,EC50),可以有效降低药物的使用量。瑞德西韦和磷酸氯喹具有低的半数细胞毒性浓度(Half-cytotoxic concentration,CC50),对人体正常细胞危害较小,瑞德西韦和磷酸氯喹的药物选择性更高,法匹拉韦对新型冠状病毒的特异性治疗效果较差[17]。
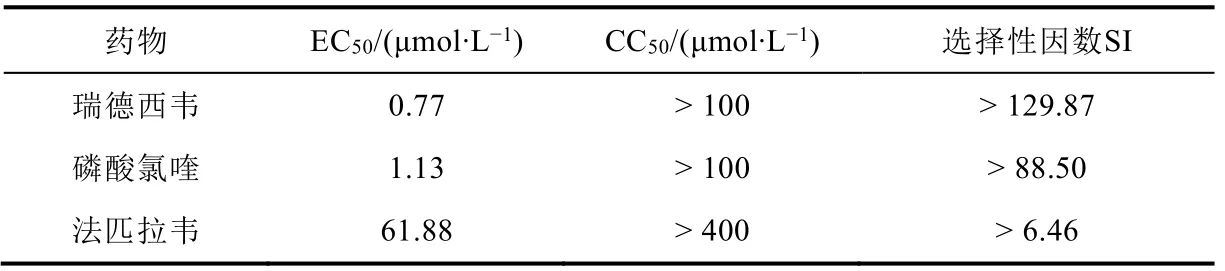
表1 磷酸氯喹和瑞德西韦药物相关参数比较
1.4 国产药物研究进展——以普鲁克胺为例
普鲁克胺(Proxalutamide,30,图10)是开拓药业自主研发的新一代雄激素受体(Androgen Receptor,AR)拮抗剂和降解剂,具有竞争性抑制AR和下调AR表达的双重机制,对新型冠状病毒感染早期和中后期均有很好的疗效,已获得巴拉圭国家公共卫生和社会福利部的紧急使用授权。

图10 普鲁克胺的结构
2020年2月,开拓药业与苏州大学收集了1339例新型冠状病毒患者使用普鲁克胺的临床数据,分析发现病毒通过将自身的刺突蛋白与人体细胞上的血管紧张素转化酶2(Angiotensin converting enzyme 2,ACE2)受体结合完成入侵,人体细胞中跨膜丝氨酸蛋白酶2(Transmembrane protease serine 2,TMPRSS2)能激活病毒的刺突蛋白。普克鲁胺能下调ACE2和TMPRSS2表达,抑制体内新型冠状病毒进一步感染其他正常宿主细胞。普鲁克胺目前处于III期临床试验(NCT04869228),已有的临床数据表明,其可以有效解决新型冠状病毒患者各阶段的感染情况[18]。除普鲁克胺外,已有多个国产抗新型冠状病毒药物,如VV116等进入研究阶段。
2 抗艾滋病病毒药物
艾滋病,即获得性免疫缺陷综合征(Acquired immunodeficiency syndrome,AIDS),是由人类免疫缺陷病毒(Human immunodeficiency virus,HIV,艾滋病病毒)引起的一种致死率很高的恶性传染病[19]。艾滋病病毒的类脂包膜来自宿主细胞,并嵌有病毒蛋白gp120与gp41。其中gp120位于表面,gp41是跨膜蛋白,与gp41通过非共价作用结合。目前,针对艾滋病的治疗方法多为高效联合抗逆转录病毒治疗,俗称“鸡尾酒疗法”,抗艾滋病病毒药物联合使用有效抑制了艾滋病病毒增殖。抗艾滋病病毒药物主要有六类,分别为核苷类逆转录抑制剂、非核苷类逆转录酶抑制剂、蛋白酶抑制剂、整合酶抑制剂、融合酶抑制剂和辅助受体拮抗剂。虽然艾滋病治疗效果不断提高,但抗艾滋病毒药物研发仍面临无法治愈与预防疾病、药物长效性不足等挑战。近两年上市的阿兹夫定作为双靶点抗艾滋病病毒药物进一步降低了艾滋病患者的用药成本,卡博努瓦作为新一代“鸡尾酒疗法”药物有效降低了用药频率,福替沙韦作为病毒附着类药物为抗艾滋病病毒药物的研发提供了新的思路。
2.1 阿兹夫定
阿兹夫定(Azvudine,31,图11)是由河南真实生物科技有限公司和郑州大学、河南师范大学、北京协和药厂等共同研发的全球首个艾滋病毒逆转录酶与Vif(Virion infectivity factor,病毒感染因子)辅助蛋白双靶点抑制剂,是国内第一个拥有自主知识产权的抗艾滋病毒口服药物,2021年7月21日由国家药品监督管理局批准上市。
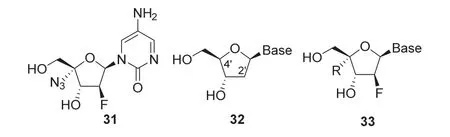
图11 阿兹夫定及其发现过程关键中间体结构
2.1.1 药物发现过程
阿兹夫定是由2’-脱氧核苷(32,图11)修饰得到的抗艾滋病药物。研究发现抗药性HIV可以从病毒前体DNA中截取2’,3’-二脱氧核苷结构片段,保留3’-OH实现抗病毒活性。进一步的研究发现,核苷的糖部分用电子缺陷基团修饰可以影响电子性质并诱导构象变化,显著提高活性。4’-叠氮基的引入可以使呋喃糖构象转变,提高抗病毒活性和叠氮胸苷的磷酸化效率。由于4’-取代-2’-脱氧核苷(4’-substituted-2’-deoxynucleosides,4’-sdN)不能用于病毒DNA生物合成,研究人员设计了新的4’-sdN(33,图11),通过对4’-位和碱基筛选,发现未取代碱基对抗病毒活性十分重要。2’-β-氟原子有效提高了药物的稳定性,特别是酸稳定性。基于以上过程,成功研发了阿兹夫定[20]。
2.1.2 药物作用机制
如图12所示,作为艾滋病病毒逆转录酶和辅助蛋白Vif双靶点抑制剂,阿兹夫定在体内有两种抑制HIV机制,一种是在HIV逆转录过程中实现抑制,阻断HIV遗传物质的合成;另一种是在HIV组装过程中,通过阻断Vif蛋白和APOBEC3G(一种胞嘧啶脱氨酶,能使逆转录病毒HIV-1基因组发生G → A的碱基突变,抑制HIV复制过程)蛋白结合,此时释放的HIV没有感染性。
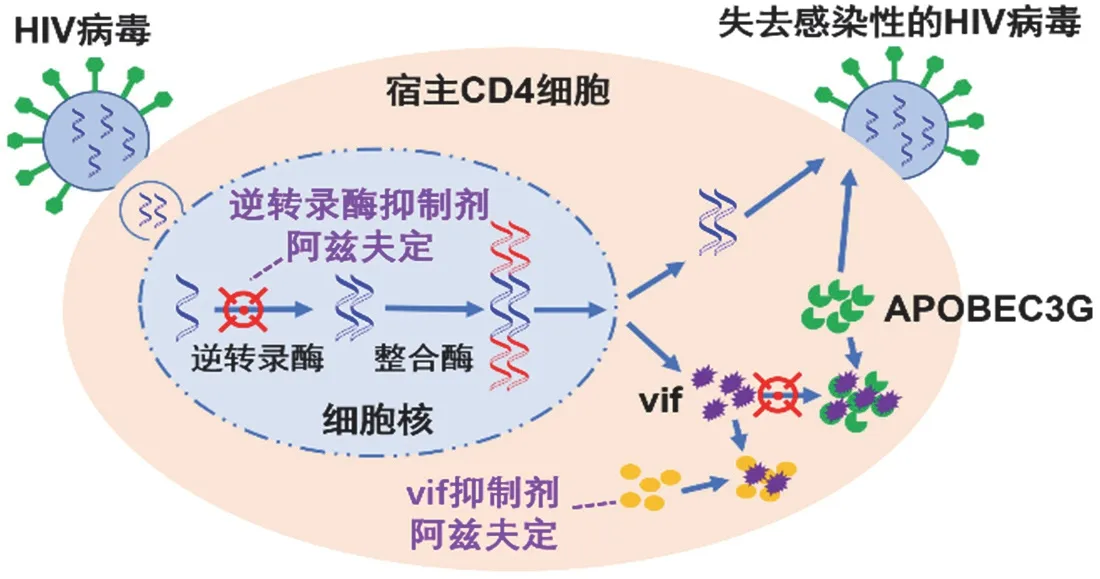
图12 阿兹夫定抗HIV机制
常俊标教授团队的抗新型冠状病毒药物筛选和临床试验发现,阿兹夫定具有一定的抗病毒活性。阿兹夫定的抗病毒作用机制与瑞德西韦和莫努匹韦类似,都是通过特异性抑制病毒RdRp活性,阻断病毒RNA的转录异常实现抗病毒[21]。
2.1.3 构效关系
阿兹夫定的主要构效关系包括:(a)用其他核碱或三唑取代胞苷会降低抗HBV活性;(b)2’-β-氟原子可以提高核苷的稳定性;(c)3’-OH基团对于抗HIV活性很重要,该位点修饰会导致抑制活性显著降低,3’-OH基团还能够降低交叉耐药/耐药性的风险;(d)4’-叠氮基不影响核苷作为延迟DNA链终止剂的能力;(e)与其他核苷一样,5’-OH基团是磷酸化的特定位点;(f)胞苷结构的5-位修饰通常导致活性降低,N4胞苷的修饰可调节抗病毒效力、毒性和其他生物特性[20]。
2.1.4 类似药物比较
在阿兹夫定上市之前,拉米夫定(Lamivudine,3TC,34,图13)和齐多夫定(Zidovudine,AZT,35)是治疗艾滋病的常用药。拉米夫定是目前临床应用疗效最好、最具代表性的核苷类似物。齐多夫定是第一个获得美国FDA批准的抗艾滋病药物,因其疗效确切,成为“鸡尾酒疗法”最基本的组合成分,在体外对逆转病毒包括HIV具有高活性。阿兹夫定、拉米夫定和齐多夫定在C8166细胞中对不同类型HIV病毒的抑制活性见表2,阿兹夫定对野生型HIV-1IIIB和HIV-1RF显示出强抑制活性,EC50为30–110 pmol∙L-1[22]。

表2 31、34和35对C8166细胞RT耐药株的抗HIV-1活性a[23]

图13 三种抗艾滋病毒药物的结构
2.1.5 阿兹夫定的合成
如图14所示,1,3,5-O-三苯甲酰基-2-脱氧-2-氟-D-阿拉伯呋喃糖苷(36)与HBr-HOAc作用产生37,其与尿嘧啶偶联得到化合物38,脱除保护基得到核苷39。39用I2/Ph3P处理,然后在甲醇钠作用下发生消除反应得到41,41与ICl/NaN3发生亲电加成反应得到42,进而发生酯化、脱保护和成盐得到阿兹夫定[23]。
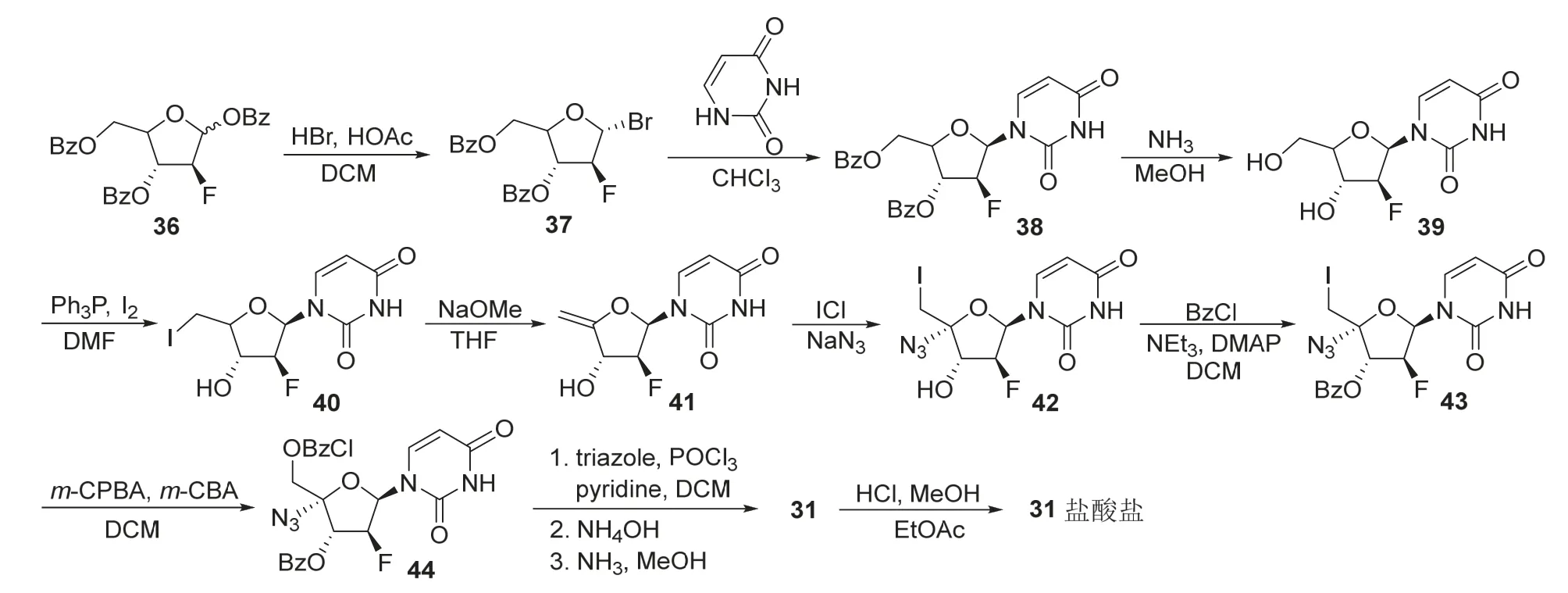
图14 一种阿兹夫定的合成路线
2.2 卡博努瓦
卡博努瓦(Cabenuva)是由英国ViiV Healthcare公司申请,2021年1月21日美国FDA批准上市的艾滋病治疗药物,用于治疗成人HIV-1感染。该药是两种可注射药物卡博特韦(Cabotegravir,45,图15)和利匹韦林(Rilpivirine,46,图15)的联合制剂,每月给药一次。作为替代当前抗HIV治疗方案的一种选择,可用于在稳定治疗方案下病毒学上受到抑制(HIV-1 RNA每毫升少于50个拷贝)、无既往治疗失败史,且对卡博特韦和利匹韦林均无已知或疑似耐药性的患者。
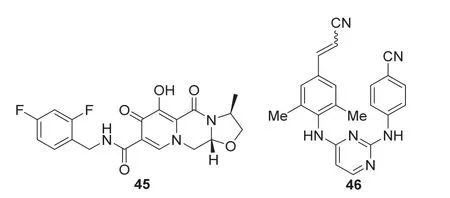
图15 卡博特韦和利匹韦林的结构
2.2.1 卡博特韦
卡博特韦是由ViiV Healthcare公司研发的第2代整合酶链转移抑制剂,用于AIDS治疗和暴露前预防,具有半衰期长、抗病毒能力强及水溶性低等优势。如图16所示,卡博特韦有两个结构特征,可以与整合酶活性位点结合:在活性位点附近的疏水口袋内结合的取代苄基和配位两个Mg2+离子形成螯合物,实现药物与活性位点相互作用[24]。其合成路线如图17所示,首先“一锅法”四步反应得到4-吡啶酮衍生物51,之后经过酰胺化反应、脱保护、环化反应得到卡博特韦[24]。
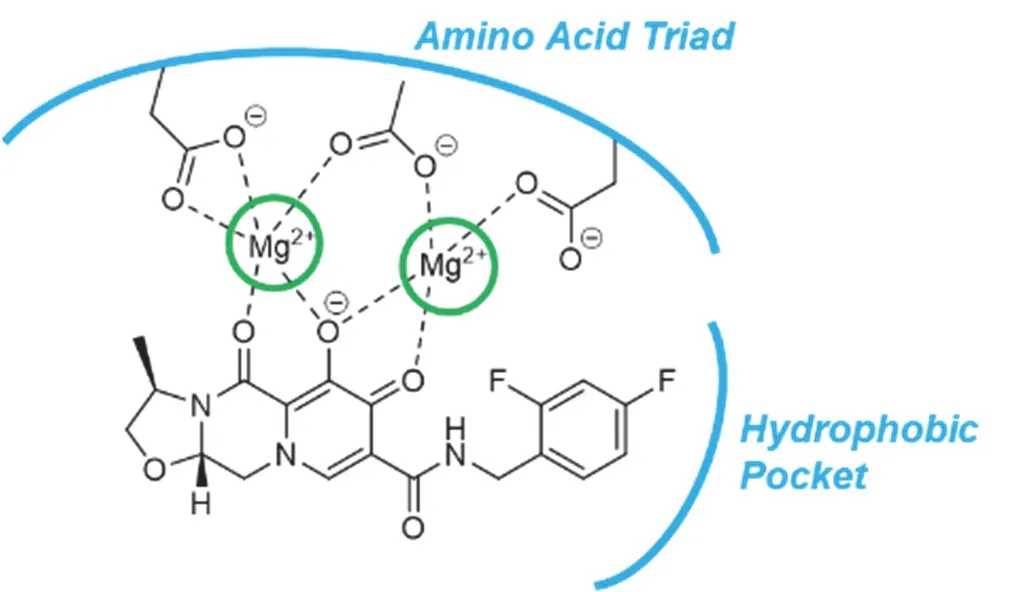
图16 卡博特韦作为整合酶抑制剂的作用机制
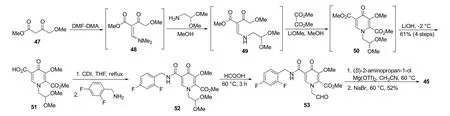
图17 一种合成卡博特韦的路线
2.2.2 利匹韦林
利匹韦林(Rilpivirine,46,图15)由Janssen公司研发,2011年5月在美国上市。适用于治疗成年1型人类免疫缺陷病毒HIV-1感染的初治患者,与其他抗逆转录病毒药物联用。利匹韦林片剂2018年11月在中国上市。
构效关系研究表明[25],利匹韦林分子中苯环2,6-位是单甲基和未取代衍生物对野生型病毒有较高效力,失去疏水作用和π–π相互作用会降低对于突变型病毒的效果。苯环6-位引入乙基或异丙基对活性影响较小,取代基的空间位阻与活性成反比,而取代基的电子效应对活性影响不大。
利匹韦林的合成如图18所示,化合物54与3,5-二甲基-4-氨基苯甲酸乙酯发生取代反应得到化合物55,经还原和氧化得到化合物57,最后发生Wittig-Wadsworth-Emmons反应得到利匹韦林[26]。

图18 一种合成利匹韦林的路线
2.2.3 药物作用机制
卡博特韦和利匹韦林都是整合酶抑制剂,HIV的RNA逆转录首先得到病毒DNA,该DNA在整合酶作用下整合到宿主细胞DNA,实现新RNA的合成。卡博特韦和利匹韦林通过抑制整合过程中的整合酶达到抗病毒效果。
2.2.4 药物治疗方案的特点
传统的艾滋病病毒治疗方案是高效联合抗逆转录病毒治疗(俗称“鸡尾酒疗法”),即针对HIV复制的三种酶——逆转录酶、整合酶和蛋白酶,通过三种或三种以上抗病毒药物联合使用,避免了单一用药产生的抗药性。该治疗方法需要每天口服药物,患者的药物依从性差。卡博努瓦是首个每月注射一次的药物,极大地方便了患者。
2.3 福替沙韦
福替沙韦(Fostemsavir,商品名:Rukobia,58,图19)是英国ViiV Healthcare公司开发的抗逆转录病毒药物,是HIV病毒附着抑制剂,用于治疗成人HIV-1感染,2020年7月FDA批准上市。

图19 福替沙韦、先导化合物和前体化合物的结构
2.3.1 药物修饰过程
福替沙韦的先导化合物是吲哚乙醛酰胺(59,图19),化合物59仅对HIV-1有抑制性,对其他病毒没有显著活性。构效关系表明,用杂环取代苯环,只有亲脂性类似物有活性;前体化合物60的7-位1,2,4-三唑改善了药代动力学性质。通过膦酰氧基修饰和与氨基丁三醇成盐得到福替沙韦[27]。
2.3.2 药物作用机制
如图20所示,正常情况下,病毒蛋白质外壳上的gp120蛋白在与宿主细胞接触前通过形态改变,暴露CD4连接活性位点实现与宿主细胞膜上CD4蛋白结合,实现病毒侵入。而作为HIV病毒附着抑制剂,福替沙韦直接与病毒包膜表面的gp120蛋白结合,使其形态不能变化,无法暴露活性位点,阻止了HIV病毒对宿主细胞膜上CD4蛋白的识别和HIV附着[28]。
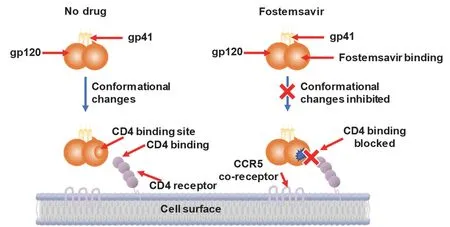
图20 福替沙韦的作用机制
2.3.3 药物构效关系
先导化合物构效关系研究发现,氮杂吲哚4-、7-位取代基不宜采用位阻大且吸电子基团,如-OiPr,-NO2等。氮杂吲哚1-位氮上只能是氢原子,取代后活性降低。哌嗪环可以修饰,但需保留六元环结构,哌嗪环2-位小位阻取代基可以增强活性。酰胺中的苯环仅能用亲脂性的杂环替代[26]。氮杂吲哚7-位杂环取代时,需要保证杂环和氮杂吲哚6-位氮具有相互作用以稳定构象,但引入的杂环2’-位可能与吲哚1-位N―H键存在相互作用,阻止了氮杂吲哚1,3-烯丙位张力作用导致的结构变化[28]。
2.3.4 合成路线
福替沙韦的第一种合成方法如图21所示[16],3-甲基-1H-1,2,4-三唑(62)和63在铜催化下偶联生成64,64和2-氯-2-氧代乙酸甲酯(65)发生Friedel-Crafts反应得到66,碱性水解得到67。然后,67在缩合剂1-(3-二甲基氨基丙基)-3-乙基碳二亚胺(EDC)存在下与苯基(哌嗪-1-基)甲酮(68)缩合得到69。最后,69与70发生亲核取代反应,再用三氟乙酸脱保护得到福替沙韦。
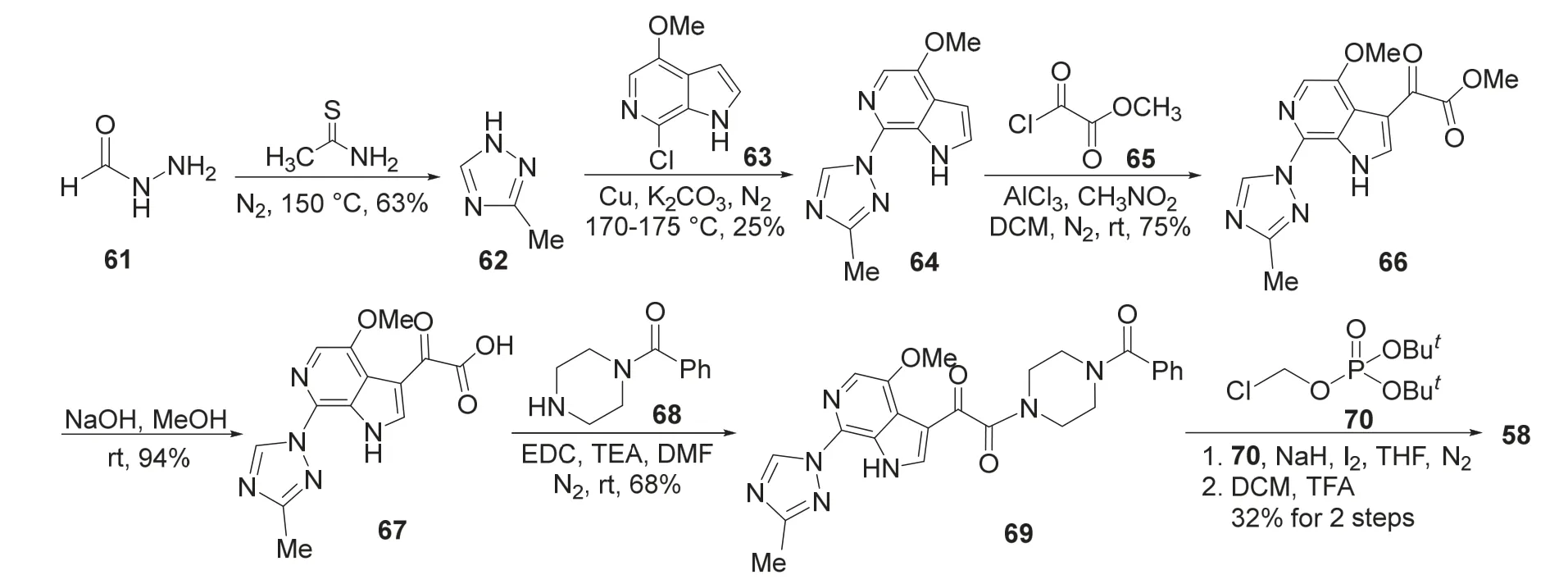
图21 一种合成福替沙韦的路线
3 抗流感病毒药物
流感是由流感病毒引起的急性上呼吸道疾病。流感病毒一般是一种单股、负链RNA病毒。流感病毒感染宿主细胞后的复制速度非常迅速,其生命周期大体可分为吸附、内吞、融合、复制、翻译、装配、出芽和释放8个环节。目前,对抗流感病毒的策略有疫苗和小分子抗流感药物。疫苗每年都需要重新配置,以适应抗原变异,其研发周期长、成本高,这些缺点使得小分子药物成为防治流感的主要手段。已经上市和处于临床阶段的抗病毒小分子药物主要包括特异性流感病毒抑制剂和广谱抗病毒药物。近期抗流感病毒药物集中在聚合酶三聚体抑制剂研究,巴洛沙韦玛波西酯(Baloxavir Marboxil,71,商品名:Xofluza,图22)是药效十分显著的抗流感病毒新药[29]。
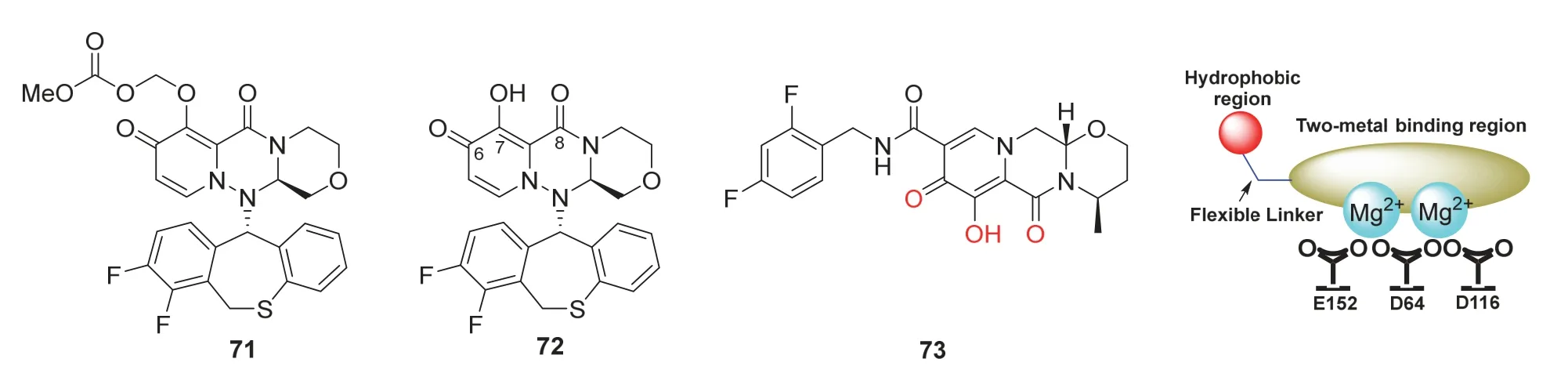
图22 巴洛沙韦玛波西酯和巴洛沙韦酸的结构及作用靶点
巴洛沙韦玛波西酯是由日本盐野义制药(Shionogi)与瑞士罗氏共同研发的一种针对甲型、乙型流感病毒的新型抗流感药物。在国内用于治疗12周岁及以上急性无并发症的流感患者,包括存在流感并发症的高风险患者。在日本获批用于治疗甲型、乙型流感病毒感染;在美国、新加坡、泰国、中国香港及台湾等地用于症状出现不超过48小时的12岁及以上急性无并发症的流感患者。巴洛沙韦玛波西酯在体内的活性物质是巴洛沙韦酸(Baloxavir acid,72,图22)。2018年2月,首先在日本获批上市。同年10月,FDA接受巴洛沙韦酯的新药上市申请,该药是近20年在美国首个获批的新型作用机制的抗流感药物。2021年4月,巴洛沙韦片获批在国内上市。
3.1 药物发现过程
巴洛沙韦玛波西酯的开发基于盐野义公司早期合成的Dolutegravir(73,图22)[30],他们参考Dolutegravir抑制HIV整合酶的关键结构,通过巴洛沙韦酸的7-位羟基和6,8-位两个羰基与2个镁离子形成螯合结构,实现了对端粒(Centromer,CEN)蛋白的抑制。之后,换用二苯并硫䓬结构单元与CEN的氨基酸残基的范德华相互作用,进一步提高了抗流感效果[31]。借助细胞表型筛选,优化化学结构,得到前药巴洛沙韦玛波西酯,提高了巴洛沙韦酸在体内的吸收[32]。
3.2 药物作用机理
由酸性聚合酶(Acidic polymerase,PA)、碱性聚合酶1(Basic polymerase 1,PB1)和2(Basic polymerase 2,PB2)组成的RNA聚合酶在流感病毒复制和转录过程中起着至关重要的作用[33]。病毒转录mRNA取决于独特的“抢帽”机制[34]。如图23所示,巴洛沙韦玛波西酯在体内水解为巴洛沙韦酸,选择性抑制帽依赖性核酸内切酶(一种参与流感病毒mRNA起始合成的关键酶),流感病毒无法完成正常转录、翻译过程。
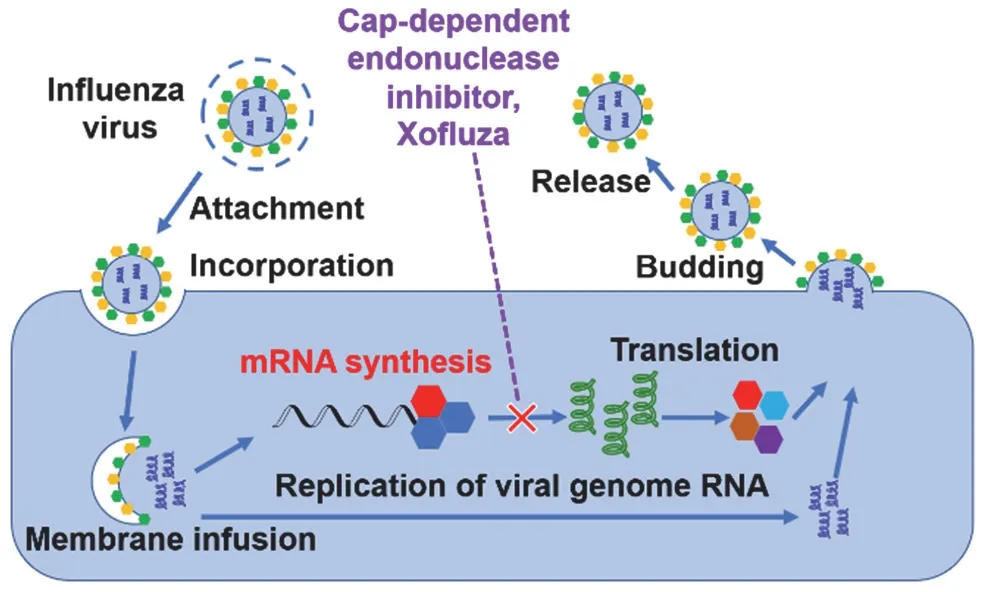
图23 巴洛沙韦玛波西酯体内抑制流感病毒机制
3.3 巴洛沙韦玛波西酯的合成
巴洛沙韦玛波西酯的合成路线如图24所示,3-苄氧基-4-氧代-4H-吡喃-2-甲酸(74)经酯化、肼解得到化合物76。N-羟乙基邻苯二甲酰亚胺(77)经成醚和脱保护得到化合物79。化合物76在DBU作用下与79发生胺解反应得到80,随后在甲基磺酸作用下成环得到外消旋体81。使用R-四氢呋喃-2-羧酸(82)对81进行手性拆分,得到手性产物83,83在碱性条件下水解得到84,84与85发生缩合反应,脱去保护基,再与氯甲基碳酸甲酯成醚得到巴洛沙韦玛波西酯[29]。
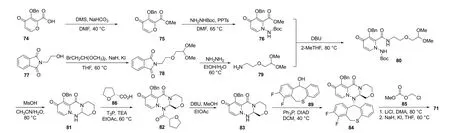
图24 一种合成巴洛沙韦玛波西酯的方法
4 总结与展望
目前,各种病毒感染疾病给人类生命健康带来了巨大挑战。截至2023年1月,全球累计确诊新型冠状病毒感染病例超过6亿6851万例,死亡病例超过673万例。虽然已经上市了新型冠状病毒疫苗和药物,但病毒快速变异仍是当前疫情防控的难点,寻找病毒新的作用靶点,并以此为基础进行药物研发是当前的研究热点之一。上市的抗新型冠状病毒药物,如莫努匹韦、帕克斯洛维德等,其作用人群有明显局限,较多药物相互作用更限制了其应用,药物也有较大副作用。此外,大多数药物用于治愈感染人群,但尚未有治疗感染新型冠状病毒治愈人群后遗症的药物。因此,研究准确识别新型冠状病毒的特异性药物和后遗症药物可能是未来新型冠状病毒药物的研发方向之一。艾滋病是一个全球公共卫生问题,目前已造成近4010万人死亡,虽然已经有较多的药物和药物联合使用治疗取得了较好的效果,但仍然没有彻底治愈的方法。当前,对于艾滋病毒储存库以及耐药性等方面,也没有较好的应对手段,已经成为攻克艾滋病的难题。病毒性流行感冒虽被人们广泛认识,但由于流感病毒变异速度快,对药物的耐药性提升,对新型抗流感药物的需求也更加迫切[35]。
近三十年来,抗病毒药物研究,特别是对抗慢性病毒感染方面已经取得了多项重大突破性进展。截至2020年,获批的抗病毒药物中艾滋病药物数量最多。但新型冠状病毒疫情表明,现有抗病毒药物严重不足,我国更是在抗病毒药物研发方面落后于西方国家,抗病毒药物有待继续深入研究。相信随着科技水平的不断进步和新的药物靶点的不断发现,人类必将研制出更多高效、副作用小的抗病毒药物。
猜你喜欢
中国预防兽医学报(2022年8期)2022-11-05
汉语世界(The World of Chinese)(2022年3期)2022-06-15
药学研究(2022年3期)2022-04-07
汉语世界(The World of Chinese)(2021年1期)2021-02-22
汉语世界(The World of Chinese)(2020年5期)2020-11-02
汉语世界(The World of Chinese)(2020年4期)2020-08-14
广东药科大学学报(2020年3期)2020-03-03
中国感染与化疗杂志(2020年6期)2020-01-12
第二课堂(小学版)(2019年4期)2019-05-13
小学生·多元智能大王(2015年7期)2015-07-03
