
改性蒙脱石负载V2O5脱除气态Hg0的性能研究*
2024-01-22 08:21:46陆克平王钧伟张先龙
煤炭转化 2024年1期
杨 森 张 楠 魏 坤 王 沙 陆克平 王钧伟 张先龙
(1.中国石油化工股份有限公司安庆分公司发展规划部,246002 安徽安庆;2.安庆师范大学化学化工学院,246011 安徽安庆;3.中国石油化工股份有限公司安庆分公司质管中心,246002 安徽安庆;4.合肥工业大学化学与化工学院,230009 合肥)
0 引 言
汞是煤中含有的有害元素,在煤燃烧过程中会释放进入烟气,由于其挥发性高、毒性具有持久性和生物累积性,会对生态环境和人体健康造成严重危害[1-2]。燃煤引起的汞污染问题受到全球的关注,各国政府先后制定相关法律法规来限制燃煤烟气中汞的排放。全球128个国家地区共同签署的首个汞限排国际公约《关于汞的水俣公约》已经正式生效[3]。我国政府高度重视燃煤汞污染,新施行的《火电厂大气污染物排放标准》进一步收紧了燃煤电厂烟气中汞及其化合物的排放限值。燃煤烟气汞排放污染问题的高效经济治理,已成为煤炭清洁利用的重要方面之一[4]。
蒙脱石是一种天然的二八面体结构的硅酸盐矿石,具有很强的离子交换性、吸水性、膨胀性、黏结性和热稳定性,经酸改性后,蒙脱石具有发达的孔隙结构、较高的比表面积和孔体积及孔径,而且具有良好的吸附性能,可以作为一种良好的吸附剂和催化剂载体。文献报道和本课题组前期研究结果表明,V2O5对Hg0具有较高的氧化活性,可将Hg0氧化为较易脱除的Hg2+[10-11]。
本研究通过负载V2O5到H2SO4改性后的MMT,制备了V2O5/MMT催化剂并将其用于脱除模拟烟气中的Hg0。希望结合活性组分V2O5的催化氧化活性和改性后MMT载体的吸附性能,将Hg0经V2O5/MMT催化剂催化氧化后生成较易脱除的Hg2+并吸附在催化剂上,实现燃煤烟气中汞的经济高效脱除。研究了H2SO4改性、催化剂制备条件、反应温度、烟气成分(O2,SO2,NO,H2O(g))等对V2O5/MMT催化剂脱除Hg0的影响,并研究了脱除Hg0后V2O5/MMT催化剂的再生性能,采用XRD,BET,XPS,SEM-EDS等方法对脱除Hg0前后的V2O5/MMT催化剂进行了表征,探讨了Hg0在V2O5/MMT催化剂上的脱除过程。
1 实验部分
1.1 催化剂的制备
称取100 g钠基蒙脱石,将其浸渍于1 000 mL质量分数为10%的H2SO4溶液中,于80 ℃搅拌4 h后抽滤、洗涤,直至最后一滴滤液呈中性后,在110 ℃烘箱中烘干12 h,即制得酸化蒙脱石,命名为10S-MMT(表示用质量分数为10%H2SO4改性后的MMT)。
将一定量的H2C2O4和NH4VO3置于50 mL去离子水中,加热搅拌使得溶液从棕黄色变至深蓝色后停止搅拌,冷却至室温后转移至100 mL容量瓶中定容,即制得V2O5前驱体溶液。
称取20 g 10S-MMT,以固液比为1 g∶1 mL的比例浸渍于上述V2O5前驱体溶液中后搅拌成泥状,自然晾干24 h,再置于烘箱中50 ℃下干燥5 h,110 ℃下干燥5 h,最后将其研磨成粒径范围为0.300 mm~0.850 mm的颗粒,置于马弗炉中,空气气氛500 ℃下煅烧5 h,即制得10S-Vx/MMT-500催化剂(Vx表示负载了质量分数为x%的V2O5(理论计算量);500表示煅烧温度为500 ℃)。
1.2 Hg0的脱除实验
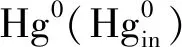
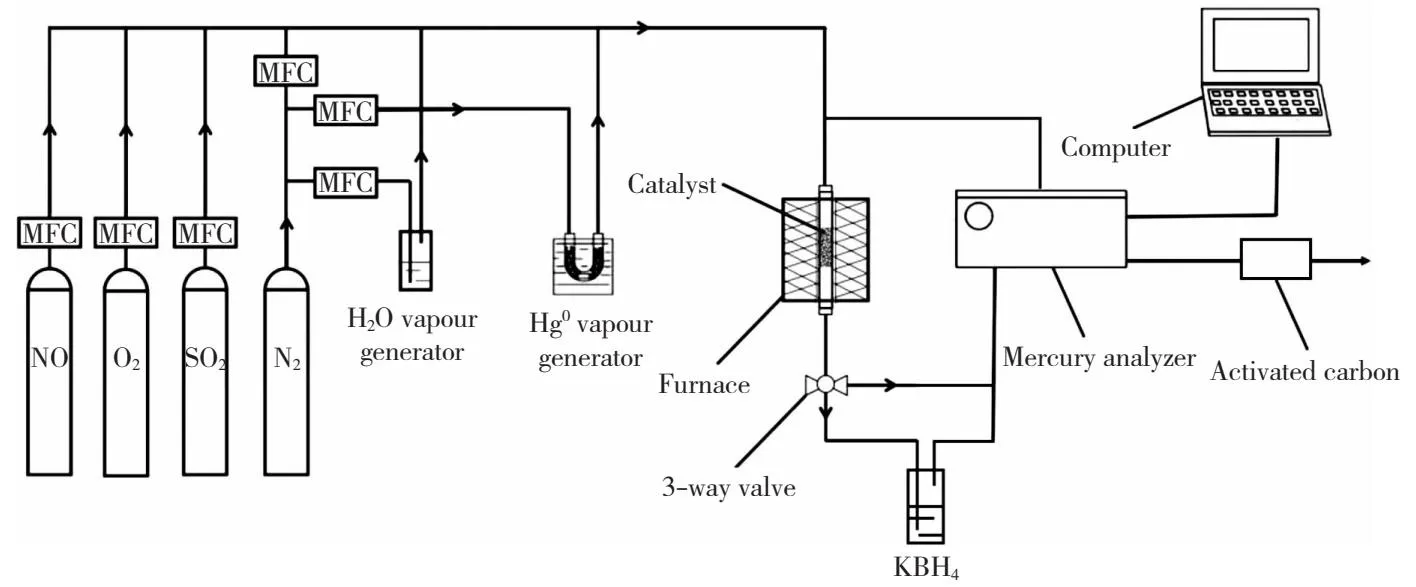
图1 Hg0脱除实验装置
(1)
1.3 催化剂的再生实验
10S-Vx/MMT-500催化剂脱除Hg0后,在图1所示的固定床反应装置对其进行热再生。再生条件为:气体体积流量为100 mL/min的N2气氛下,程序升温(升温速率为10 ℃/min)至再生温度400 ℃并保持恒温再生2 h,然后切换至空气气氛中在300 ℃下继续加热2 h。再生后的10S-Vx/MMT-500催化剂再次在模拟烟气中进行脱除Hg0的实验。为进一步考察10S-Vx/MMT-500催化剂的使用寿命,对10S-Vx/MMT-500催化剂进行了多次脱除Hg0-再生-脱除Hg0的实验[2],再生后的催化剂标记为Rn-10S-Vx/MMT-500(n表示催化剂再生次数)。
1.4 程序升温脱附实验
程序升温脱附(temperature programmed desorption,TPD)实验在图1中所示的固定床反应装置中进行。样品为在模拟烟气(N2+5%O2+0.15%SO2+0.05%NO+5%H2O+230 μg/mHg0)中150 ℃下脱除Hg0后的10S-V3/MMT催化剂,加入量约为10 mg。样品加入后,通入氮气吹扫30 min,然后以10 ℃/min的升温速率由室温升至800 ℃,反应器出口气体经KBH4溶液还原后通入测汞仪在线连续检测其中汞的质量浓度。
1.5 催化剂的表征
1.5.1 BET测试
采用ASAP2460型多站扩展式全自动比表面与孔隙度分析仪(美国麦克仪器公司生产)测定V2O5/MMT催化剂的比表面积、孔体积和孔径分布。催化剂的比表面积采用BET方法进行计算,孔径分布采用BET方程和N2吸附等温线的脱附曲线计算。
1.5.2 扫描电子显微镜能谱(SEM-EDS)
采用ZEISS Sigma 300型场发射扫描电镜(德国蔡司公司生产)观察催化剂的外观形貌和催化剂表面的元素分布。
1.5.3 X射线光电子能谱(XPS)
使用单色AlK α辐射作为放射源在ESCALAB250型X射线光电子能谱仪(美国ThermoScientic公司生产)上测量研究10S-Vx/MMT-500催化剂表面电荷效应,以C1s=284.6 eV为标准进行荷电校准。
1.5.4 X射线衍射(XRD)
X射线衍射分析在XRD-7000 S X射线衍射仪(日本岛津株式会社生产)上进行,使用CuK α射线辐射,扫描范围10°~80°,扫描速度为5°/min,工作电压和电流分别为40 kV和100 mA。
2 结果与讨论
2.1 催化剂的表征结果
2.1.1 XRD表征结果
采用XRD谱表征了改性前MMT与10%H2SO4改性后MMT的结构变化,结果如图2所示。由图2可以看出,未改性的MMT样品出现了六种物质的衍射峰,其中2θ为20.03°,28.05°和31.31°的衍射峰归属于MMT(PDF#13-0259);2θ为21.96°和36.25°的衍射峰归属于方石英(PDF#27-0605);2θ为21.26°,26.08°和29.55°的衍射峰归属于石英(PDF#46-1045);2θ为13.87°的衍射峰归属于沸石(PDF#15-0179);2θ为23.82°的衍射峰归属于青金石(PDF#41-1393);2θ为25.98°的衍射峰归属于柱沸石(PDF#39-1381)。其中2θ为7.49°的衍射峰归属于MMT镜面001;2θ为18.26°的衍射峰归属于MMT镜面020[12]。对于H2SO4改性后的MMT样品,MMT镜面001衍射峰发生了偏移,2θ从7.49°偏移至9.22°,且衍射峰的强度下降。通过Bragg公式算出MMT镜面001的层间距d001从1.18 nm(MMT)降低至0.962 5 nm(10S-MMT),这可能是因为MMT是由一层铝氧八面体和两层硅氧四面体组成,通过H2SO4改性,溶液中的H+不断置换出铝氧八面体中的阳离子,使得MMT结构中晶体结构被破坏,层间距降低[13]。未改性MMT镜面020的衍射峰变化并不明显,但是改性后在2θ为18.26°处出现了MMT镜面020衍射峰,这主要是因为未改性的MMT表面被大量的杂质覆盖,H2SO4改性可腐蚀杂质,使得被杂质覆盖的MMT镜面020暴露出来[14-15]。
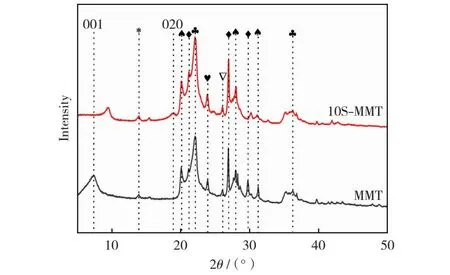
图2 MMT和10S-MMT的XRD谱
10S-MMT-500及不同V2O5负载量的10S-V1/MMT-500,10S-V3/MMT-500和10S-V5/MMT-500催化剂的XRD表征分析结果如图3所示。由图3可以看出,不同V2O5负载量的三种催化剂的XRD谱中并未发现明显的V2O5衍射峰,这可能是由于活性组分V2O5在MMT载体表面分散性很好,因此没有检测到V2O5衍射峰[16]。
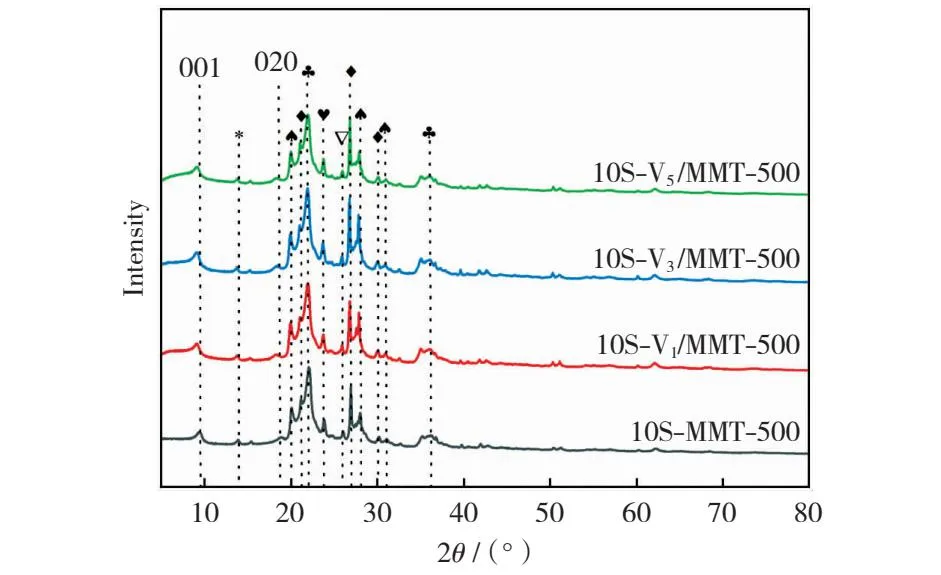
图3 10S-MMT-500和10S-Vx/MMT-500的XRD谱
2.1.2 BET表征
MMT,10S-MMT-500,10S-V1/MMT-500,10S-V3/MMT-500和10S-V5/MMT-500及R5-10S-V3/MMT-500催化剂的BET表征结果如表1所示。由表1可以看出,经10%H2SO4改性后MMT的比表面积大幅度增大,从36.57 m2/g增大至62.63 m2/g,且孔体积也有所增大,这与XRD的表征结果(见图2)一致。对于V2O5/MMT催化剂,随着V2O5负载量增大,催化剂的比表面积和孔体积均明显下降,5%V2O5负载量的V2O5/MMT的比表面积和孔容分别降低至14.36 m2/g和0.050 cm3/g。这是因为当载体负载活性组分V2O5后,V2O5分布在载体的孔隙之间,在一定程度上堵塞了孔道[17]。

表1 催化剂的BET表征
2.1.3 SEM-EDS表征
MMT,10S-MMT-500和10S-V3/MMT-500催化剂的SEM照片如图4所示。由图4可以看出,未改性MMT的结构较为完整,表面呈现一些层状结构。经过10%H2SO4改性后的MMT表面结构完整性被破坏,片状结构增多,孔隙结构增多。10S-V3/MMT-500催化剂表面的片状结构与10S-MMT-500催化剂表面的片状结构相比未发生明显改变,片状边缘发生轻微蜷曲,且堆积方式更为杂乱[18]。

图4 MMT,10S-MMT-500和10S-V3/MMT-500的SEM照片
10S-V3/MMT-500催化剂表面V元素的mapping分析如图5所示。由图5可以看出,活性组分V2O5已经成功负载到MMT载体上,而且在载体MMT表面的分散性很好,这与XRD的表征结果(见图3)是一致的。V2O5在载体MMT表面的均匀分散,有利于其对Hg0的氧化。

图5 10S-V3/MMT-500表面V元素的mapping分析
2.2 V2O5负载量对10S-Vx/MMT-500脱除Hg0的影响
V2O5负载量对10S-Vx/MMT-500脱除Hg0的影响如图6所示。由图6可以看出,未改性的MMT几乎没有脱除Hg0的能力。经10%H2SO4改性后,10S-MMT-500脱除Hg0的能力有所提高,Hg0的脱除效率提升至50%左右。10S-Vx/MMT-500催化剂脱除Hg0的能力明显高于MMT和10S-MMT-500脱除Hg0的能力,Hg0的脱除效率达到90%以上。其中,V2O5负载量为3%的10S-V3/MMT-500催化剂脱除Hg0的能力最高,Hg0的脱除效率达到98%以上。继续增加V2O5的负载量至5%并没有进一步提高Hg0的脱除效率,这主要是由于负载量为3%的V2O5已经可以很好地氧化Hg0,继续增加V2O5的负载量会堵塞催化剂孔道,影响了Hg0在催化剂上的吸附和氧化,导致Hg0的脱除效率降低[19-20]。
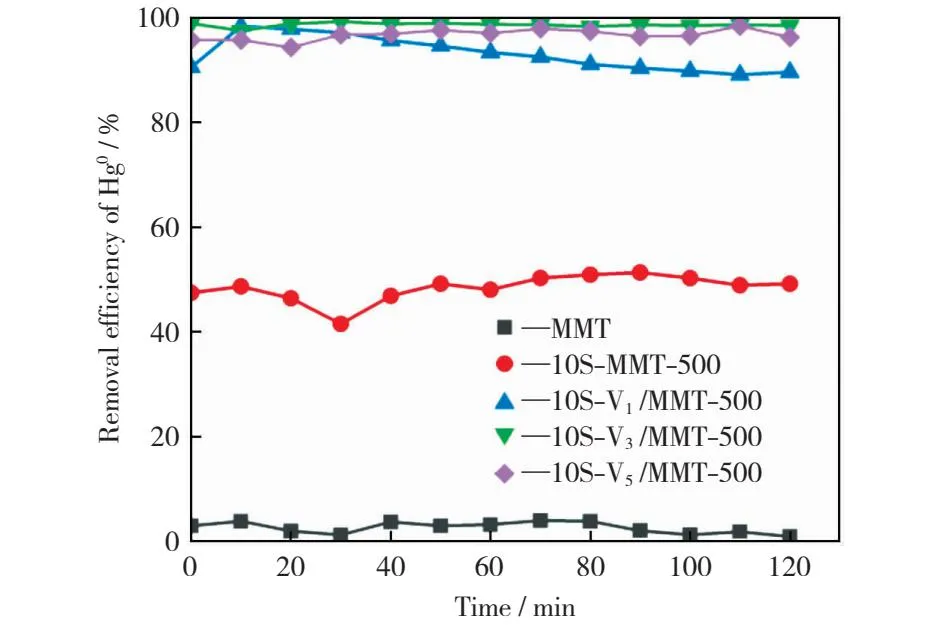
图6 MMT,10S-MMT和10S-Vx/MMT脱除Hg0的能力对比
2.3 温度对10S-V3/MMT-500脱除Hg0的影响
10S-V3/MMT-500催化剂在不同反应温度(120 ℃,150 ℃,180 ℃和210 ℃)下Hg0的脱除效率对比如图7所示。由图7可以看出,当反应温度从120 ℃升高至150 ℃时,10S-V3/MMT-500脱除Hg0的能力提高,继续升高温度至210 ℃,10S-V3/MMT-500脱除Hg0的能力降低。这主要是因为不同温度对Hg0在催化剂上的吸附和氧化影响不同,低温有利于Hg0的吸附但不利于Hg0的氧化,而高温有利于Hg0的氧化但不利于Hg0的吸附。150 ℃时Hg0的吸附和氧化的共同作用最好,使得10S-V3/MMT-500具有最高的脱除Hg0的能力。
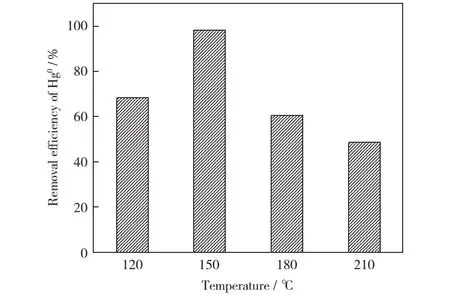
图7 温度对10S-V3/MMT-500脱除Hg0的影响
2.4 烟气成分对10S-V3/MMT-500脱除Hg0的影响
10S-V3/MMT-500催化剂在N2,N2+5%O2,N2+0.15%SO2,N2+0.15%SO2+5%O2,N2+0.05%NO和N2+5%H2O(g)等六种不同气氛中Hg0脱除效率对比如图8所示。由图8可以看出,10S-V3/MMT-500催化剂在N2气氛中就具有良好的脱除Hg0的能力,4 h时Hg0的脱除效率达到65.7%。当N2中加入体积分数为5%的O2后,催化剂脱除Hg0的能力明显提高,4 h时Hg0的脱除效率升高至78.2%。O2对Hg0的脱除具有促进作用,可能主要是因为O2能够补充10S-V3/MMT-500氧化Hg0过程中消耗的晶格氧,也可以吸附在催化剂表面形成吸附氧,有利于Hg0的氧化,这与文献[21]中报道的O2对金属氧化物催化剂脱除Hg0的影响作用是一致的。

图8 烟气成分对10S-V3/MMT-500脱除Hg0的影响
文献[22-23]中报道的SO2对V2O5基催化剂脱除Hg0的影响作用差别很大,本研究也考察了SO2对10S-V3/MMT-500催化剂脱除Hg0的影响。由图8可以看出,气氛中加入体积分数为0.15%的SO2后,Hg0的脱除效率有所升高,特别是同时加入体积分数为0.15%的SO2和体积分数为5%的O2后,Hg0的脱除效率提高至98%以上,明显高于N2+5%O2气氛中Hg0的脱除效率。由于V2O5对Hg0和SO2均有氧化作用,SO2的加入会与Hg0竞争V2O5活性位,从而会导致10S-V3/MMT-500对Hg0的脱除效率下降。但实验结果却与此相反,这表明Hg0和SO2在10S-V3/MMT-500上并不只是简单存在竞争吸附关系,SO2和O2在V2O5的催化作用下会生成SO3,生成的SO3可与Hg0反应生成HgSO4,从而提高了催化剂脱除Hg0的能力,这与课题组前期的研究结果和文献[24-26]中报道的结果是一致的。
NO对10S-V3/MMT-500催化剂脱除Hg0具有促进作用。由图8可以看出,当N2中加入体积分数为0.05%的NO后,Hg0的脱除效率明显提高至94%以上,这是因为NO可以在10S-V3/MMT-500表面形成NO2等物质,可促进Hg0的氧化[21]。
H2O(g)对10S-V3/MMT-500催化剂脱除Hg0具有抑制作用。由图8可以看出,N2中加入体积分数为5%的H2O(g)后,Hg0的脱除效率有所下降,从65.7%下降至55.6%。这主要是因为H2O(g)会吸附在催化剂表面占据活性位,与Hg0存在竞争吸附,影响了Hg0在催化剂活性位上的吸附和氧化[27]。
2.5 催化剂上吸附Hg的形态
脱除Hg0前后的10S-V3/MMT-500催化剂上Hg 4f和V 2p的XPS谱如图9所示。由图9可以看出,对于脱除Hg0前后的10S-V3/MMT-500催化剂,均在结合能为102.5 eV左右出现了1个峰,这归属于载体SiO2中的Si 2p峰[28],脱除Hg0后的10S-V3/MMT-500催化剂上并没有发现Hg 4f的衍射峰,这主要是因为Hg 4f的衍射峰与Si 2p的衍射峰结合能区间重合程度较大[29],而且催化剂上吸附Hg的质量远小于载体中Si的质量,因此没有发现Hg 4f的衍射峰。脱除Hg0前的10S-V3/MMT-500催化剂在结合能为517.3 eV处出现了V5+的峰[30],表明脱除Hg0前催化剂表面的活性组分主要是以V2O5的形式存在的,而对于脱除Hg0后的10S-V3/MMT-500催化剂,在结合能为517.5 eV和516.2 eV处分别出现了V5+和V4+的峰,经计算n(V5+)/n(V4++V5+)为76.6%。这表明10S-V3/MMT-500催化剂在脱除Hg0的过程中,部分V5+转化成了V4+,证实了V2O5对Hg0的氧化活性在10S-V3/MMT-500脱除Hg0过程中的关键作用。

图9 脱除Hg0前后10S-V3/MMT-500上Hg与V的XPS谱
采用程序升温脱附实验进一步研究了10S-V3/MMT-500催化剂上吸附Hg的形态,结果如图10所示。由图10可以看出,对于脱除Hg0后的10S-V3/MMT-500催化剂,在温度从20 ℃升温至800 ℃的过程中出现了3个Hg的释放峰,Hg的释放从150 ℃左右开始,在200 ℃,305 ℃和650 ℃左右的峰分别归属于Hg0,HgO和HgSO4的释放峰[31],这证实了催化剂上存在3种不同形态的Hg,即Hg0,HgO和HgSO4。从3个Hg的释放峰的面积可以看出,HgO是Hg主要存在的形态,进一步证实了V2O5对Hg0的氧化活性在10S-V3/MMT-500脱除Hg0过程中的关键作用。Hg0被V2O5氧化生成了HgO和HgSO4并吸附在10S-V3/MMT-500催化剂上。TPD的实验结果也解释了图7中的结果,即不同温度下10S-V3/MMT-500催化剂对Hg0的脱除是氧化和吸附共同作用的结果。

图10 脱除Hg0后10S-V3/MMT-500上Hg的程序升温脱附结果
2.6 10S-V3/MMT-500催化剂的再生
为了考察10S-V3/MMT-500催化剂的再生性能,对10S-V3/MMT-500催化剂进行了多次脱除Hg0-再生-脱除Hg0的实验,结果如图11所示。由图11可以看出,经过多次脱除Hg0-再生实验后的10S-V3/MMT-500催化剂仍然具有良好的脱除Hg0的能力。第1次再生后的10S-V3/MMT-500催化剂的Hg0脱除效率在120 min时达到80%左右;第5次再生后的10S-V3/MMT-500催化剂的Hg0脱除效率在120 min时仍达到60%左右。随着脱除Hg0-再生次数的增加,再生后催化剂脱除Hg0的能力有所下降,这可能主要有两个原因所致:一是由于10S-V3/MMT-500催化剂在400 ℃下多次加热再生,载体MMT会发生部分孔道破坏塌陷(见表1中再生后催化剂的比表面积和孔结构数据),导致催化剂孔道结构发生变化和部分活性组分流失;二是由于10S-V3/MMT-500催化剂在400 ℃下再生,并未能将催化剂上吸附的Hg全部释放出来,HgSO4和部分HgO仍存在于催化剂上占据活性位(见图10中HgSO4和HgO的释放温度区)。虽然多次再生后10S-V3/MMT-500催化剂脱除Hg0的能力有所下降,但再生5次后的催化剂的Hg0脱除效率仍高于60%,而且远高于MMT脱除Hg0的能力,这表明10S-V3/MMT-500催化剂具有良好的再生循环使用性能。
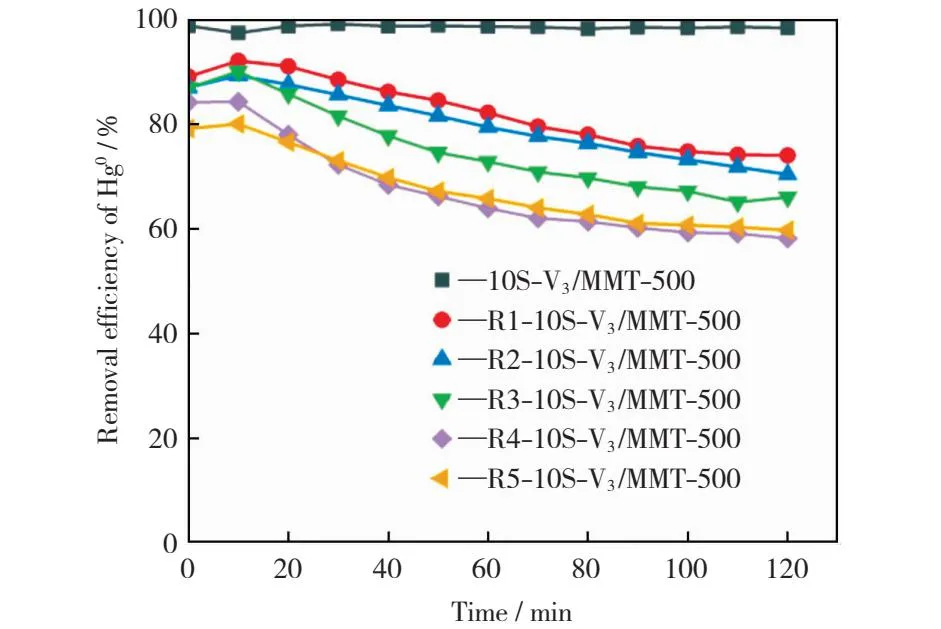
图11 10S-V3/MMT-500的再生性能
3 结 论
1) 质量分数为10%的H2SO4改性可使MMT载体具有更高的比表面积和更发达的孔道结构,同时增强了其对Hg0的吸附能力。
2) V2O5/MMT催化剂具有良好的脱除Hg0的活性和稳定性。V2O5负载量对10S-V2O5/MMT脱除Hg0的能力具有明显影响,负载量为3%的V2O5的10S-V3/MMT-500催化剂在反应温度为150 ℃、体积空速为6 000/h条件下对Hg0的脱除效率达到98%以上。
3) O2和NO对10S-V3/MMT-500催化剂脱除Hg0具有促进作用;SO2对10S-V3/MMT-500催化剂脱除Hg0具有一定的促进作用;H2O(g)对10S-V3/MMT-500催化剂脱除Hg0具有抑制作用。
4) Hg0在10S-V3/MMT-500上被氧化生成了含Hg2+的化合物HgO和HgSO4并吸附在10S-V3/MMT-500上。
5) 脱除Hg0后的10S-V3/MMT-500催化剂再生后,仍具有良好的脱除Hg0的能力。V2O5/MMT催化剂具有良好的再生循环使用性能。
猜你喜欢
化学工程师(2023年1期)2023-02-17 15:09:48
化工管理(2022年13期)2022-12-02 09:21:52
理化检验-化学分册(2020年12期)2021-01-26 00:41:38
中国果树(2020年2期)2020-07-25 02:14:28
上海农业科技(2019年1期)2019-02-22 01:51:28
测控技术(2018年2期)2018-12-09 09:00:52
中国塑料(2016年12期)2016-06-15 20:30:07
中国资源综合利用(2016年2期)2016-01-22 07:27:41
中国塑料(2015年11期)2015-10-14 01:14:14
中国塑料(2015年9期)2015-10-14 01:12:17
