
基于硼亲和分子印迹策略的MOF/MIPs对沙丁胺醇的选择性吸附和计算模拟
2024-01-18 02:52:12王鼎南张宜明
高等学校化学学报 2024年1期
李 欣, 周 颖, 王鼎南, 裴 勇, 武 斌, 张宜明
(1. 浙江农林大学食品与健康学院, 杭州 311300;2. 浙江省水产推广技术总站, 杭州 310023; 3. 湘潭大学化学学院, 湘潭 411105;4. 浙江农林大学数学与计算机科学学院, 杭州 311300)
沙丁胺醇(Salbutamol, SAL)是在治疗哮喘和慢性阻塞性肺疾病中使用最广泛的β2激动剂之一[1].沙丁胺醇对牛、 羊、 禽和猪等具有促生长、 促进营养再分配及提高动物蛋白质合成等作用, 所以饲料中添加沙丁胺醇可以显著提高肉类行业的利润[2,3]. 但是, 人在食用含沙丁胺醇较高的动物组织后将产生一系列不良反应(如肌肉震颤、 心悸、 头痛、 恶心和呕吐等), 会对身体健康造成重大危害[4]. 因此, 动物产品中沙丁胺醇残留物的检测和测定对畜牧业和公共卫生健康都具有重要意义. 目前, 沙丁胺醇残留检测的方法[弱阳离子交换层析、 聚合物微萃取、 免疫亲和柱和固相萃取(SPE)柱等[5~7]]很多, 但这些方法设备昂贵且操作繁琐、 选择性差.
分子印迹聚合物(Molecularly imprinted polymers, MIPs)是以待测物质为模板合成的超分子聚合材料, 随着新印迹技术的发展, 其对特定的目标分子(如药物、 食品和农药)具有越来越好的选择性和亲和力, 并已被用于分离和识别广泛的生化实体[8,9]. 但是MIPs的缺点(电导率差、 腔体可达性差和传质速率低)限制了其对分析物的高效检测[10], 因此, 设计具有良好导电性和高比表面积的新型MIP材料来克服这些缺陷具有重要意义. 研究表明, 金属有机框架(Metal-organic frameworks, MOFs)材料具有高孔隙率和高比表面积等优点, 是开发固定单体和模板分子的新型分子印迹聚合物的良好载体[11]. MOFMIPs 的组合结构既能提供选择性吸附的特异性识别位点, 又能提供较多的比表面积孔隙吸附[12]. 因此, 具有增强传感特性的MOF-MIPs已经被制造出来并用于检测各种目标物[13,14]. 此外, 含有特定官能团配体的MOFs被设计合成出来, 拓展了其应用[15], 近年来, 具有硼酸官能团的配体已被成功组装, 得到的功能化MOFs对顺式二醇化合物具有较好的选择性和富集性能[16,17], 硼酸配体与含顺式二醇化合物之间形成的硼酸酯可在不同的pH 下实现成环和开环的过程, 从而实现模板底物的结合和释放[18].同时, 分子印迹所构筑的选择性空腔能进一步保证硼亲和材料对含有顺式二醇结构分子的选择性能,实现快速、 稳定、 有选择性地富集食品中的含顺式二醇的污染物质.
近年来, 为了提高MIPs的可预测性、 可控性、 选择性和识别能力, 并定量探索印迹分子与功能单体之间的相互作用, 已有在原子水平上建立分子模型模拟MIPs的设计[19]. 目前, 分子模拟在分子印迹材料的研发过程中得到了越来越广泛的应用, Zhao等[20]对甲醛和甲基丙烯酸复合物自组装系统中交联剂、 溶剂以及印迹比的优化模拟, 为制备快速、 经济、 新型的甲醛MIPs 提供了可靠的理论依据.Zeng等[21]用密度泛函理论分析了泰乐菌素与硼酸盐以带电氢键形式存在的硼酸亲和机理. 分子模拟不仅为功能单体、 印迹比、 交联剂和溶剂[22,23]的筛选提供了理论指导, 而且定量描述了分子水平[24,25]上的识别和机制以及印迹的本质.
鉴于形成的硼酸酯键对底物分子的独特识别能力, 借助这种亲和作用可在MOF骨架结构上构筑新型微流控选择性识别芯片. 本文以Zr为金属中心, 以对苯二甲酸(Terephthalic acid, BDC)和3-羧基苯硼酸(3-Carboxyphenylboronic acid, 3-CPBA)为双配体, 合成了UiO-66-BA金属有机框架材料, 并在此基础上开发了一种基于硼亲和策略的新型MOF/MIP复合材料的分子印迹纸基芯片FSU-BA@MIP, 通过密度泛函理论(Density functional theory, DFT)对FSU-BA@MIP选择性吸附沙丁胺醇的机理进行了理论计算; 首先对FSU-BA@MIP 中具有吸附作用的金属有机框架(UiO-66-BA)的3-羧基苯硼酸配体和沙丁胺醇的复合物进行拓扑分析, 对比了中性和离子化硼酸与沙丁胺醇间的相互作用力; 再通过Materials Studio软件的DMol3模块对UiO-66-BA和沙丁胺醇及竞争吸附物质的复合物进行模拟; 计算结果解释了基于硼亲和分子印迹策略MOF/MIPs对沙丁胺醇的选择性吸附, 并为下一步的MOF基选择性识别界面构筑提供了实例分析和理论参考.
1 实验部分
1.1 计算方法
通过Gaussian 16[26]和Materials Studio 2018[27]软件进行结构优化, 使用Multifwn 3.8[28]软件分析结果, 用VMD1.9.3[29]软件进行可视化.
1.2 计算过程
1.2.1 硼亲和作用机理 采用Gaussian 16程序研究3-羧基苯硼酸配体和沙丁胺醇的相互作用, 结构优化使用隐式溶剂模型(Implicit solvation model, SMD), 溶剂为水, 采用DFT的B3LYP/6-31+G(d,p)泛函加上色散校正(em=gd3bj)计算, 最后用波函数分析软件Multifwn进行拓扑分析, 并用VMD软件绘制结构模型.
1.2.2 印迹空腔模拟 印迹空腔的模拟能够预测分子印迹空穴的形状, 使用Materials Studio 软件的Adsorption Locator 模块的模拟退火法获得了3-CPBA 和模板沙丁胺醇分子以及交联剂硅酸四乙酯(Tetraethyl orthosilicate, TEOS)的自组装混合物, 再将模板分子从中移除, 形成含有印迹空腔的混合物, 然后对其进行几何优化, 并用Forcite模块的动力学方法模拟了腔体对模板再吸附的动态过程.
1.2.3 印迹空腔识别机制 对沙丁胺醇和几种竞争吸附化合物[去甲肾上腺素(Noradrenaline,NORE)、 特布他林(Terbutaline sulfate, TER)、 克仑特罗(Clenbuterol hydrochloride, CBL)、 莱克多巴胺(Ractopamine hydrochloride, RAC)]进行结构优化, 再使用Multifwn 软件分析得出每种底物分子的体积. Piletsky等[30]提出只有模板分子或分子体积小于模板分子的物质才能够进入印迹空腔, 在此, 通过比较分子体积的大小初步确定能够进入印迹空腔的化合物, 由此来判断分子印迹对竞争化合物是否存在吸附作用.
1.2.4 结合能与反应能 使用Materials Studio 软件中的DMol3模块进行选择性吸附计算, 具体模拟过程如下: (1) 对构建好的UiO-66-BA、 沙丁胺醇、 竞争吸附化合物进行结构优化; (2) 在已经优化完的UiO-66-BA吸附SAL的xsd文件去除模板(SAL)后固定的所有原子坐标, 然后在可能的作用位点加入优化完成的竞争化合物形成新的复合构型; (3) 根据结合能公式计算得出几种物质与印迹空腔中识别位点的结合能, 通过比较结合能的大小说明模板对沙丁胺醇的特异性吸附. 并通过Mulliken布居电荷分析能够更加准确地从理论上说明FSU-BA@MIP对沙丁胺醇的选择性吸附.
根据下式计算结合能(∆E1, eV):
式中:EC(eV)为UiO-66-BA与吸附化合物形成未成环简单复合物的总能量;ET(eV)为吸附化合物的能量;EU(eV)为UiO-66-BA的能量.
因为硼酸配体与含顺式二醇的化合物在一定的pH下可形成的硼酸酯, 故模拟硼亲和分子印迹对目标物的选择性时不能仅仅计算一般的非共价作用情况. 在此, 也模拟计算了UiO-66-BA和SAL及竞争物质反应脱去两分子水形成五元或六元环酯时的反应能, 在式(1)的基础上加上两分子水的能量得到的反应能(∆E2, eV)的公式如下:
式中:EM(eV)为UiO-66-BA与吸附化合物结合成含有硼酸环酯复合物的总能量;EH2O(eV)为水的能量.
1.3 实验过程
为了考察FSU-BA@MIP 对 SAL 的特异性吸附, 选择同属于β-肾上腺素受体激动剂的克仑特罗、 特布他林、 莱克多巴胺及结构类似物去甲肾上腺素作为SAL 的竞争吸附化合物. 分别配制上述5种化合物的标准甲醇溶液(50 mg/L, 2 mL), 加入两片FSU-BA@MIP或FSU-BA@NIP 在水浴摇床中以120 r/min 振荡1 h, 结束后取上层清液过0.22 μm 针头式滤膜, 用高效液相色谱[Agilent 1100型高效液相色谱仪(HPLC), 美国安捷伦科技有限公司]检测残留物浓度, 并根据下式计算吸附容量:
式中:Qe(mg/g)为单位质量的FSU-BA@MIP 对SAL 的吸附量;c0(mg/L)为SAL 起始溶液浓度;ce(mg/L)为吸附平衡时溶液相中SAL 浓度;V(mL)为吸附平衡时溶液体积;m(g)为FSU-BA@MIP 的质量.
2 结果与讨论
2.1 沙丁胺醇和3-羧基苯硼酸的相互作用
分子中原子(Atoms in molecules, AIM)理论分析化学键主要是基于电子密度拓扑分析的手段. AIM拓扑图中显示了键径和键临界点(Bond critical point, BCP), 从实际化学意义上判断, 键径能够较好地展现原子间相互作用的最主要路径, BCP 的属性可以用来考察相应化学键的特征, 包括强度和本质[31]. 基于 Hirshfeld 分区的独立梯度模型(IGMH)以图形的方式更直观显示分子间弱相互作用, 图形中等值面的大小和颜色可以判断作用力的强弱[32]. 在此, 可用AIM分析和IGMH图来识别3-CPBA和沙丁胺醇形成的复合物之间的相互作用.
中性的3-CPBA与沙丁胺醇分子间形成一条键径[图1(A)], 形成的等值面对应着散点图[图1(C)]中sign(λ2)ρ为-0.04处的峰值(δinterg为3-CPBA与沙丁胺醇间的相互作用力), 图1中最下面的颜色标尺代表图中的着色方式和对应的颜色解释, 所以图1(A)中形成的蓝色等值面表示强吸引力(如氢键). 离子化的3-CPBA与沙丁胺醇形成3条键径[图1(B)], 位点2的等值面对应着散点图[图1(D)] sign(λ2)ρ为-0.05处的峰值, 此处峰值明显高于位点1, 并且图1(B)中两个分子间也存在一些范德华力作用, 这些可以说明离子化的3-CPBA与沙丁胺醇间的相互作用更强. 键临界点的电子密度分析可以从理论上预测真空相中氢键的能量, 根据式(4)或式(5)[33], 可以表明实验中分子间相互作用的强度. 键临界点1处和2处的电子密度[ρ(r), a.u.]的值列于表1.

Table 1 Electron density and predicted energy H-bonding of different sites

Fig.1 Interaction of salbutamol and 3-carboxyphenylboronic acid(A) AIM and IGMH plots for neutral 3-CPBA and SAL; (B) AIM and IGMH plots of charged 3-CPBA and SAL; (C) interaction scatter plot of neutral 3-CPBA and SAL; (D) interaction scatter plot of charged 3-CPBA and SAL. Elements: H(white); B(pink);C(cyan); O(red); N(blue).
式中:Eneutral(kJ/mol)为中性复合物中形成的氢键键能. 将位点1相应的ρ(r)代入式(4), 可以计算出位点1处形成的氢键键能估计值为-35.5356 kJ/mol, 属于中等强度氢键范畴;Echarged(kJ/mol)为带电复合物中的氢键键能, 将位点2的电子密度代入式(5)得到氢键键能为-68.5631 kJ/mol, 属于强氢键. 氢键的能量越高, 共价键就越容易形成, 说明离子化3-羧基苯硼酸更容易与含顺式二醇的沙丁胺醇形成稳定的苯硼酸酯.
2.2 印迹空腔模拟
对3-CPBA, SAL和TEOS进行印迹空腔模拟, 形成的分子印迹自组装混合物如图2(A)所示, 形成的自组装混合物可以可视化模拟吸附沙丁胺醇的过程, 更为直观了解印迹空腔的形成以及其对SAL的吸附. 从图2(A)可见, 3-CPBA和SAL被TEOS包围, 形成了一定厚度的印迹涂层, Bie等[34]认为适当厚度的印迹涂层能够决定MIP对模板的结合性质, TEOS形成的二氧化硅层能够有效避免特异性吸附. 将模板沙丁胺醇分子移除, 能看到形成了一个印迹空腔[图2(B)], 由于二氧化硅的高度交联结构使印迹涂层具有高孔隙率、 优异的物理刚性等特点, 因此, 该空腔在形状和尺寸上应与模板互补. 再通过分子动力学模拟吸附沙丁胺醇的过程, 将沙丁胺醇放在空腔入口处[图2(C)], 印迹空腔里的作用位点对沙丁胺醇的特异性吸附使其进一步进入空腔, 图2(D)为分子动力学模拟结束状态.
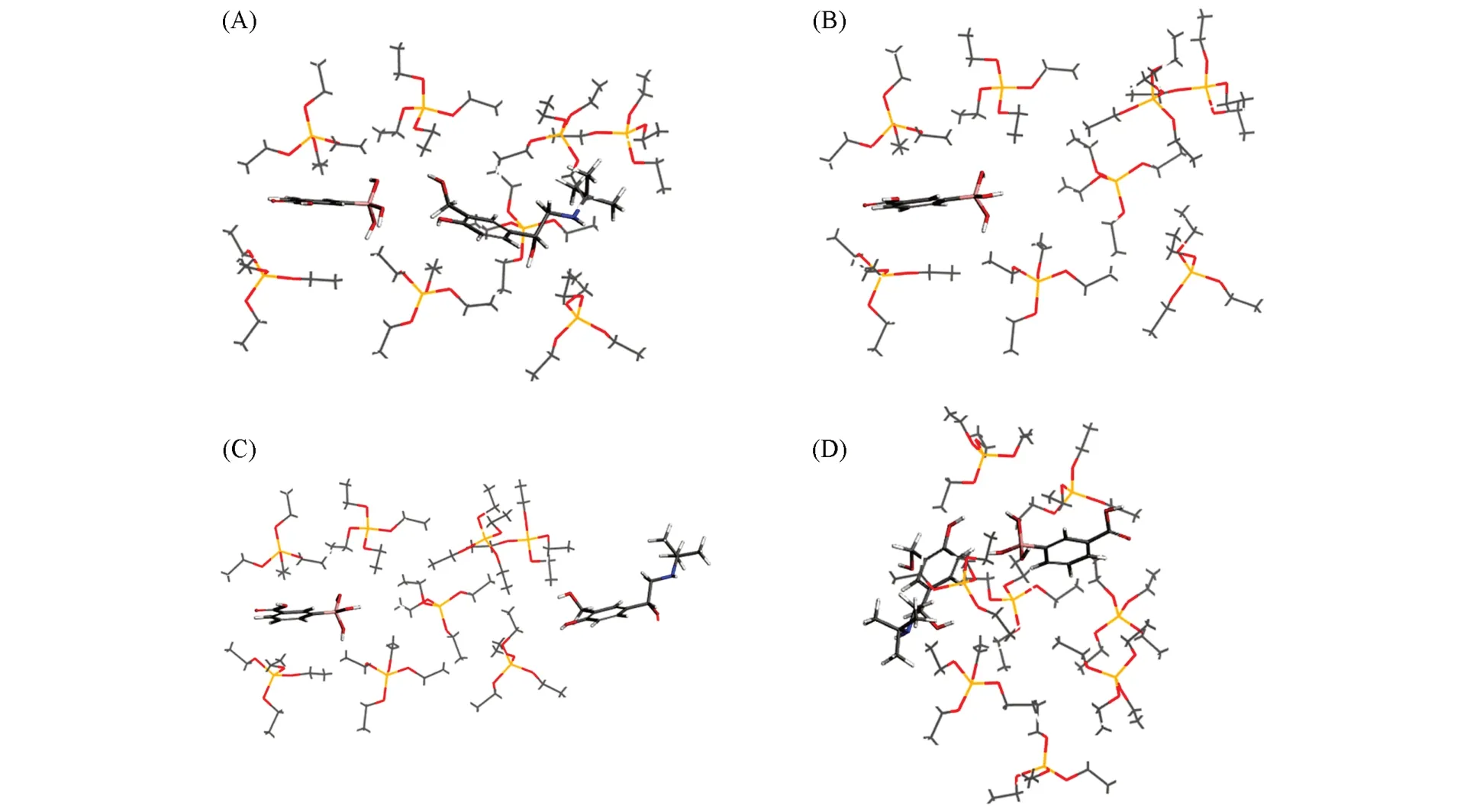
Fig.2 3-CPBA, SAL and TEOS molecularly imprinted self-assembly mixtures(A), imprinted cavity mixture(B), the initial state(C) and the end state(D) of molecular dynamics simulationElements: Si(yellow); H(white); B(pink); C(gray); N(blue); O(red).
2.3 选择性吸附模拟
研究了基于硼亲和策略新型MOF/MIP 复合材料的分子印迹吸附膜FSU-BA@MIP 对沙丁胺醇及其类似物的选择性, 实验结果表明, FSU-BA@MIP 对沙丁胺醇存在特异性吸附, 对其它竞争物质有少量吸附. 为了更加了解FSU-BA@MIP对沙丁胺醇的吸附性能并且验证实验结果, 对竞争物质的分子体积进行比较和DFT 计算, 从分子水平上对FSU-BA@MIP 中主要具有吸附作用的金属有机框架(UiO-66-BA)对沙丁胺醇的选择性识别机制进行了理论计算与分析.
2.3.1 模型的构建 探讨的硼亲和策略分子印迹技术是在纸基上进行硼亲和定向表面印迹选择性吸附动物源性食品中的沙丁胺醇, 实验中以Zr为金属中心、 对苯二甲酸和3-羧基苯硼酸为双配体(摩尔比3∶1)合成了UiO-66-BA 纳米材料. 为了让计算模拟与实验条件接近, 在UiO-66-BA 模型构建时的两种配体摩尔比也应该为3∶1, 并且Song 等[35]使用离子化4-羧基苯硼酸构型对硼酸配体与模板的结合能进行计算, 将UiO-66-BA中一个3-羧基苯硼酸配体以阴离子硼酸基形式表示, 构建的UiO-66-BA 模型如图3所示. UiO-66-BA 的结构单元是由[Zr6O4(OH)4]金属簇团与12 个配位连接而成, 密集的结构单元使得整个结构稳定地连接. 沙丁胺醇和其它竞争吸附化合物的分子构型取自ChemSpider网站.
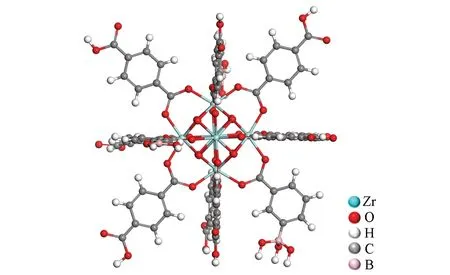
Fig.3 Initial configuration of UiO-66-BA
2.3.2 分子体积 研究分子印迹的识别机制, 首先分析竞争分子的分子体积. 使用Gaussian 16软件对沙丁胺醇及4种竞争吸附化合物[去甲肾上腺素(NORE)、 特布他林(TER)、 克仑特罗(CBL)、 莱克多巴胺(RAC)]进行结构优化, 将结果文件导入分析软件Multifwn3.8得到各自的体积.
计算得出沙丁胺醇、 克仑特罗、 莱克多巴胺的分子体积分别为0.3305, 0.3418 和0.4061 nm3, 竞争物质克仑特罗和莱克多巴胺的分子体积均大于沙丁胺醇的体积, 因此进入不了印迹空腔, 能够解释实验结果中FSU-BA@MIP对这两种物质的吸附量最低的现象. 去甲肾上腺素和特布他林的分子体积分别为0.2124和0.3068 nm3, 均比沙丁胺醇的分子体积小, 说明这两种分子可以进入印迹空腔, 但是去甲肾上腺素和特布他林分子与空腔中识别位点的相互作用弱于模板分子沙丁胺醇, 属于非特异性吸附结合, 此结论可通过DFT计算进行理论模拟验证分析.
2.3.3 UiO-66-BA与目标物质的结合能 在考虑分子印迹作用时, 印迹空腔能对模板分子沙丁胺醇及其类似物具有选择识别特性, 故UiO-66-BA 与沙丁胺醇和竞争分子去甲肾上腺素、 特布他林之间均含有相互作用力, 结合方式由目标分子和UiO-66-BA形成复合物的结构决定. 模板分子与功能单体在溶液中以非共价方式作用, 主要是氢键、 疏水作用、π-π共轭等作用力[36]. 由2.1 节的结果可知,UiO-66-BA的硼酸配体与沙丁胺醇间存在氢键作用, 在此, 先模拟UiO-66-BA与目标分子非共价作用时摩尔比为1∶1的复合物构型(图4), 并用式(1)计算结合能. 通常, 复合物的结合能越低, 复合物的稳定性越强, UiO-66-BA与目标分子之间的相互作用也越强.

Fig.4 Optimized configuration of the complex formed by UiO-66-BA with Salbutamol(A),Norepinephrine(B) and Terbutaline(C)
从表2可见, UiO-66-BA和沙丁胺醇的结合能为-1.1494 eV, 明显低于UiO-66-BA-NORE的结合能(-0.8350 eV)以及UiO-66-BA-TER 的结合能(-0.7241 eV), 结合能ΔE为负值, 越低越有利于UiO-66-BA对沙丁胺醇的识别, 说明印迹空腔对沙丁胺醇的吸附结合作用强于去甲肾上腺素和特布他林, 这与选择性吸附实验中去甲肾上腺素和特布他林的吸附量均低于沙丁胺醇的结果一致.
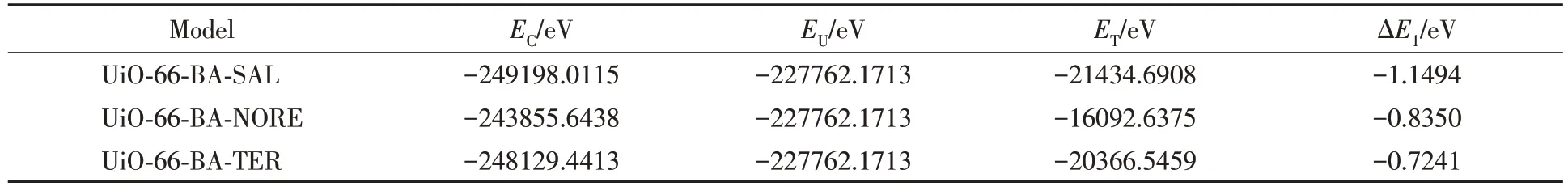
Table 2 Binding energy of UiO-66-BA with adsorbed substance
2.3.4 Mulliken布居电荷分析 Mulliken布居电荷有助于了解UiO-66-BA与沙丁胺醇、 去甲肾上腺素和特布他林形成复合物时表面电子的定量转移情况. 表3列出了3种复合物中主要原子吸附前后的电荷量, 并计算了电子转移数量(Δe), 电子转移主要是UiO-66-BA 的羟基上的O 原子得电子, H 原子失电子. 转移的电荷数越多, 相互作用越大[37], 通过表3数据可知, UiO-66-BA-SAL的得失电子数都是最多的, 因此, 在以非共价方式作用的模型中, UiO-66-BA和沙丁胺醇之间有较强的相互作用力. Mulliken电荷分析的结果和结合能的结果一致, 均能验证选择性吸附实验中FSU-BA@MIP对沙丁胺醇的吸附效果最好.
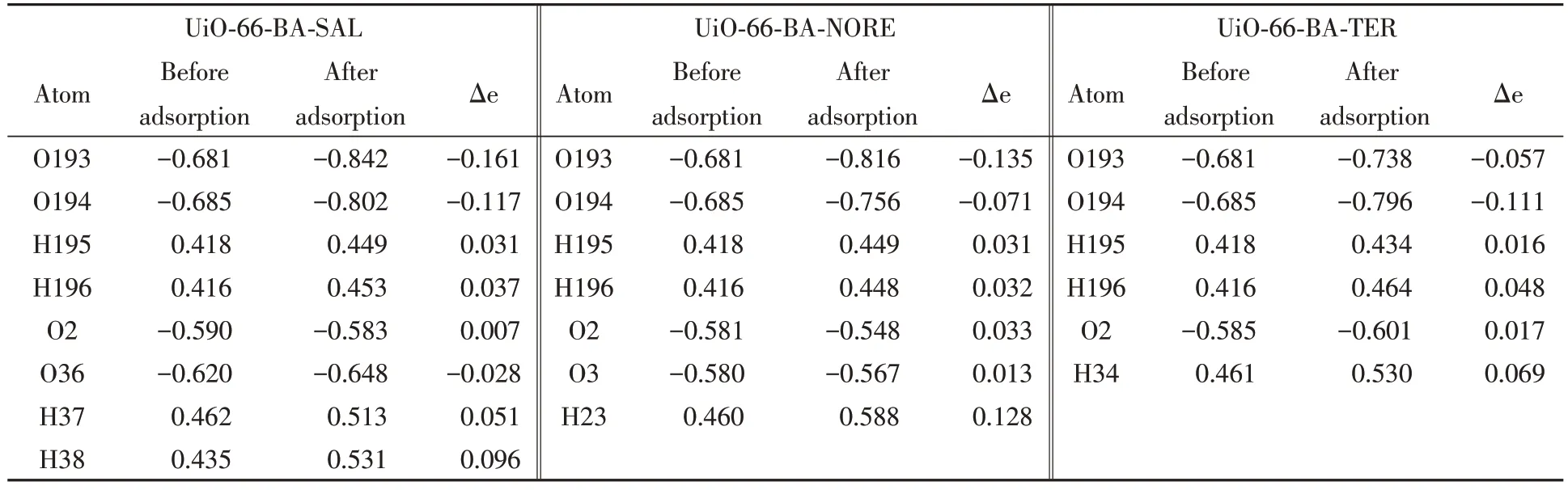
Table 3 Mulliken charge of UiO-66-BA bound to the adsorbed substance
2.3.5 UiO-66-BA与目标物质的反应能 硼酸盐亲和材料的分子相互作用的原理依赖于硼酸配体与含顺式二醇的化合物之间的可逆共价反应, 当溶液的pH大于硼酸的解离常数(pKa)值时, 硼酸以四方硼酸根阴离子(sp3)的形式存在, 并与顺式二醇反应形成共价键环酯[38]. 在模拟计算FSU-BA@MIP对沙丁胺醇选择性吸附机理时, 除了考虑分子印迹的作用, 还应考虑硼亲和作用. 使用Materials Studio 软件中的DMol3模块计算UiO-66-BA 和沙丁胺醇及竞争分子的顺势二醇反应脱去两分子水时的复合物能量, 溶剂为水, 用式(2)计算的反应能结果列于表4. 可见, UiO-66-BA与沙丁胺醇的反应能最小, 并且反应能远高于非共价作用时的能量, 能够说明UiO-66-BA的硼酸配体与SAL更易形成硼酸环酯且形成的复合物在一定pH的溶液状态下稳定存在.
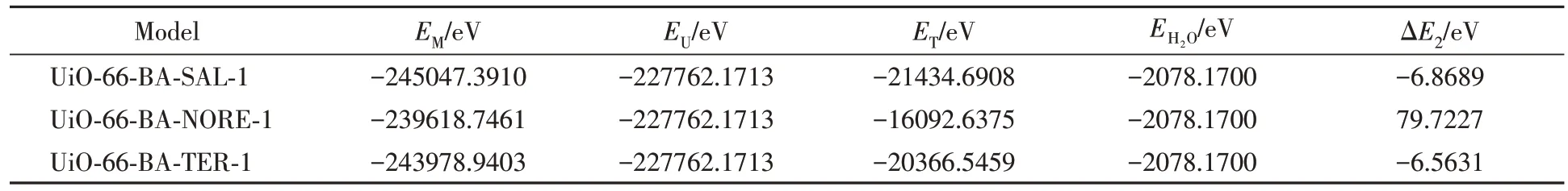
Table 4 Reaction energy of UiO-66-BA with adsorbed substance
2.4 实验结果
硼酸盐亲和材料能够在碱性环境下与含顺式二醇的物质结合; 表面印迹的有效传质促进了模板分子的去除和重新结合, 获得了较高的印迹效率. 在硼酸盐亲和与表面印迹的双重作用下,FSU-BA@MIP 具有特异性识别位点以及较高结合亲和力, 能够选择性吸附具有顺式二醇结构的沙丁胺醇, 其它相似分子存在一些非特异性吸附. 在此, 通过式(3)计算了FSU-BA@MIP 对几种物质的吸附量, FSU-BA@MIP 对沙丁胺醇的吸附量最高, 达到111.6732 mg/g, 去甲肾上腺素的吸附量为31.1263 mg/g, 高于特步他林的16.4869 mg/g, 与计算得到的结合能中去甲肾上腺素的结合能绝对值高于特步他林一致. 克伦特罗和莱克多巴胺的吸附量分别为13.8163和11.2247 mg/g, 与克仑特罗和莱克多巴胺的分子体积大于沙丁胺醇从而进入不了印迹空腔导致吸附量较低的结论一致, 说明计算结果能验证与指导实验, 并能估计选择性实验中吸附物质的吸附量高低.
3 结论
使用拓扑分析得出3-羧基苯硼酸与沙丁胺醇间存在氢键作用, 并且离子化的3-羧基苯硼酸与沙丁胺醇的氢键键能更高. 氢键的能量越高, 共价键就越容易形成, 说明硼酸以四方硼酸根阴离子(sp3)的形式存在时易与顺式二醇反应并形成硼酸酯. 此外, 通过密度泛函理论计算模拟验证实了验结果,FSU-BA@MIP对沙丁胺醇有特异性吸附, 为实验提供了计算方法与理论依据, 并为发展新型硼亲和印迹材料提供了计算参考和佐证. 对MOF界面硼亲和结合底物的计算与表征策略的研究结果, 证明了硼修饰的UiO-66-BA是一个非常具有潜力的硼亲和骨架.
猜你喜欢
大学物理(2022年9期)2022-09-28 01:10:52
装备制造技术(2021年2期)2021-07-21 05:38:08
物理通报(2020年7期)2020-07-01 09:28:02
水利科技与经济(2017年5期)2017-04-22 02:39:36
发明与创新·中学生(2017年1期)2017-01-20 20:37:52
发明与创新(2017年2期)2017-01-18 08:48:21
西南国防医药(2016年7期)2016-12-01 06:01:27
中国卫生标准管理(2015年13期)2016-01-15 02:58:22
原子与分子物理学报(2015年3期)2015-11-24 12:49:34
文理导航(2015年26期)2015-09-29 14:12:24
