
超高效液相色谱-串联质谱法快速测定化妆品中87种禁用原料
2024-01-14 13:50:44李丽霞丁晓萍李晓健
色谱 2024年1期
胡 贝, 李丽霞, 丁晓萍, 刘 红, 黄 伟, 吕 稳, 李晓健
(湖北省药品监督检验研究院, 湖北省药品质量检测与控制工程技术研究中心, 湖北 武汉 430075)
《化妆品安全技术规范》(2015年版)[1]是国家及省级化妆品监督抽检工作的检验和判定依据,其中列出的禁用原料按是否具有功效分为两类:一类在化妆品中不具有功效,且不太可能以原料或杂质的形式出现在化妆品中;另一类在化妆品中具有一定的功效,如具有美白祛斑、抗菌消炎、抗过敏、改善红血丝、舒缓助眠和增加香味等功能。目前,许多禁用原料在《化妆品安全技术规范》(2015年版)中尚无检测方法,但已有学者检出了非法添加标准方法以外的禁用原料[2-4],因此,化妆品禁用原料的筛查范围亟待扩展,筛查维度也亟需加大。
目前,化妆品中多类别禁用原料的同时检测方法主要有液相色谱-高分辨质谱法[5-7]和液相色谱-串联质谱法[8-11]。液相色谱-高分辨质谱法可以获得精确的质量数,为目标化合物乃至未知物分析提供全面的数据[12],但仪器价格昂贵,检验成本高,在方法推广上有一定难度。超高效液相色谱-串联质谱法具有特异性好、灵敏度高等优点,能快速、准确地定量化妆品中的禁用原料,在化妆品中性激素类[13-15]、抗感染类[16,17]、抗组胺类[18-19]、镇痛类[20]等药物的检测方面均有应用,是目前化妆品禁用原料检测标准方法建立和日常监督检验的首选方法。
本文基于超高效液相色谱-串联质谱,通过优化前处理和仪器条件,建立了化妆品中87种禁用原料的分析方法。87种禁用原料分别为33种性激素类、20种抗感染类、15种抗组胺类、7种香豆素类、4种镇静催眠类、4种解热镇痛类、2种致敏类药物及2种具有收缩血管作用的药物。本方法适用于3类常见化妆品基质(水基类、乳液膏霜类和油基类)的检测,可为化妆品中禁用原料的快速风险筛查和高效日常监管提供技术支持。
1 实验部分
1.1 仪器、试剂与材料
Acquity UPLC H-CLASS超高效液相色谱仪、Xevo TQ-S质谱仪(美国Waters公司); HMV-50A多管漩涡混合器(上海珂淮仪器有限公司); LC-250超声波清洗器(山东济宁鲁超超声设备有限公司); ST16台式离心机(德国Thermo Fisher Scientific公司); Milli-Q纯水机(美国Millipore公司)。
甲醇和乙腈(色谱纯,德国默克公司);正己烷(分析纯,国药集团化学试剂有限公司);实验用水为GB/T 6682中规定的一级水。87种禁用原料标准品购自中国食品药品检定研究院、德国Dr. Ehrenstorfer公司、坛墨质检标准物质中心、上海义淮生物有限公司、广州佳途科技股份有限公司、上海甄准生物科技有限公司、上海安谱璀世标准技术服务有限公司、上海源叶生物科技有限公司、天津阿尔塔科技有限公司和美国Stanford Chemicals公司,纯度为95.0%~99.9%(CAS号见表1)。
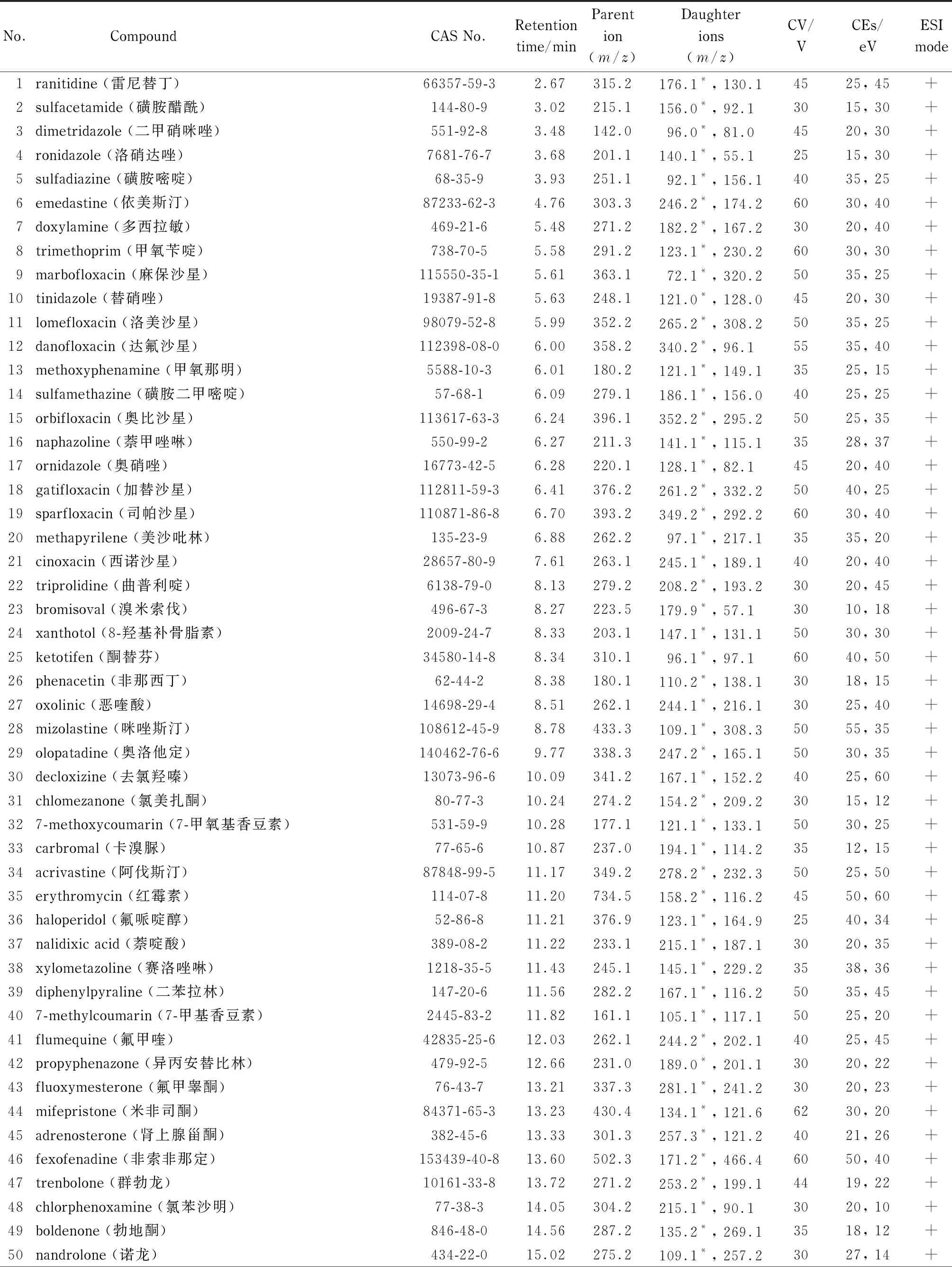
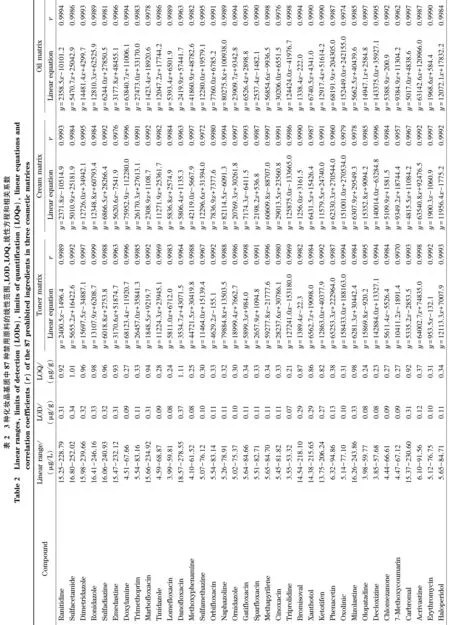
表1 87种禁用原料的CAS号、保留时间和质谱参数
1.2 标准溶液的配制
1.2.1标准储备溶液
分别准确称取10 mg标准品(精确至0.01 mg)置于10 mL容量瓶中,加入乙腈溶解(其中在乙腈中溶解较差的洛美沙星、司帕沙星、氟甲喹、麻保沙星、达氟沙星和加替沙星可以加入少量甲酸促进溶解),定容至刻度并摇匀,配制成质量浓度为1 000 mg/L的标准储备溶液,于-20 ℃下冷冻保存。
1.2.2单标溶液
使用时准确移取20 μL各标准储备溶液,分别置于不同的10 mL容量瓶中,用乙腈稀释定容并摇匀,配制成质量浓度为2 mg/L的各单标中间溶液。再准确移取1 mL各单标中间溶液分别置于不同的10 mL容量瓶中,用乙腈稀释定容并摇匀,配制成质量浓度为200 μg/L的各单标溶液。
1.2.3混合标准溶液及基质匹配标准溶液
使用时准确移取0.1 mL各标准储备溶液,置于10 mL容量瓶中,用乙腈稀释定容并摇匀,配制成质量浓度为10 mg/L的混合标准中间溶液;再准确移取1 mL混合标准中间溶液,置于10 mL容量瓶中,用乙腈稀释定容并摇匀,配制成质量浓度为1 mg/L的混合标准溶液。临用前,用空白基质提取液稀释混合标准溶液,配制成所需质量浓度的基质匹配标准溶液。
1.3 样品前处理
水基类和乳液膏霜类:称取0.2 g样品(精确至0.000 1 g)置于10 mL具塞比色管中,加入2 mL乙腈涡旋分散均匀后,再加入50%乙腈水溶液至近刻度,超声提取20 min,静置至室温;之后用50%乙腈水溶液定容至刻度,摇匀,以8 000 r/min离心5 min,取上清液过0.22 μm有机滤膜,滤液作为供试品溶液备用。
油基类:称取0.2 g样品(精确至0.000 1 g)置于15 mL具塞离心管中,加入2 mL正己烷涡旋分散均匀后,再加入3 mL 70%乙腈水溶液涡旋2 min,以8 000 r/min离心5 min;用胶头滴管移取下层溶液至10 mL具塞比色管中,再在上层正己烷层加入3 mL 70%乙腈水溶液涡旋2 min,以8 000 r/min离心5 min,用胶头滴管移取下层溶液至同一10 mL具塞比色管中,加入50%乙腈水溶液定容至刻度,摇匀,过0.22 μm有机滤膜,滤液作为供试品溶液备用。
1.4 仪器条件
1.4.1色谱条件
色谱柱:CORTECS C18色谱柱(150 mm×2.1 mm, 2.7 μm);流动相:A为乙腈,B为0.1%甲酸水溶液;柱温:40 ℃;流速:0.3 mL/min;进样体积:2 μL。梯度洗脱程序:0~2 min, 5%A; 2~4 min, 5%A~20%A; 4~13 min, 20%A~35%A; 13~16 min, 35%A; 16~18 min, 35%A~45%A; 18~26 min, 45%A~90%A; 26~31 min, 90%A; 31~31.1 min, 90%A~5%A; 31.1~33 min, 5%A。
1.4.2质谱条件
离子源:电喷雾电离(ESI)源,正、负离子扫描;检测方式:多反应监测(MRM)模式;毛细管电压:3.2 kV(正离子)和3.0 kV(负离子);脱溶剂气流速:900 L/h;脱溶剂气温度:450 ℃;锥孔气流速:150 L/h。其他质谱参数见表1。
2 结果与讨论
2.1 UPLC-MS/MS条件优化
2.1.1质谱条件的优化
采用电喷雾电离,分别将87种禁用原料的单标溶液(200 μg/L)注入离子源,在正、负离子扫描模式下对目标化合物进行一级质谱全扫描。正离子模式下,目标化合物均产生[M+H]+准分子离子峰;负离子模式下,目标化合物均产生[M-H]-准分子离子峰。确定准分子离子峰后对锥孔电压(CV)进行优化,再进行二级质谱扫描,选取丰度较高且干扰较小的2个子离子分别作为定性、定量离子,其中响应较高的离子作为定量离子。再分别优化2个子离子的碰撞能量(CE),经优化后得到的质谱参数见表1。
2.1.2色谱条件的优化
根据相关文献[5,6,8-11]及国家标准[1],同时分离激素类、抗感染类、抗组胺类和解热镇痛类药物等多个类别禁用原料的流动相体系中,水相多采用含有不同体积分数的甲酸水溶液。本研究考察了0.1%甲酸水溶液和甲醇、0.1%甲酸水溶液和乙腈、0.1%甲酸水溶液和0.1%甲酸乙腈3种流动相体系对待测物峰形及响应的影响。结果显示,在流动相体系为0.1%甲酸水溶液和乙腈的条件下,87种目标物的响应最好,且87种禁用原料中母离子和子离子质荷比相同或相近的组分均可得到分离,87种禁用原料的总离子流色谱图见图1。
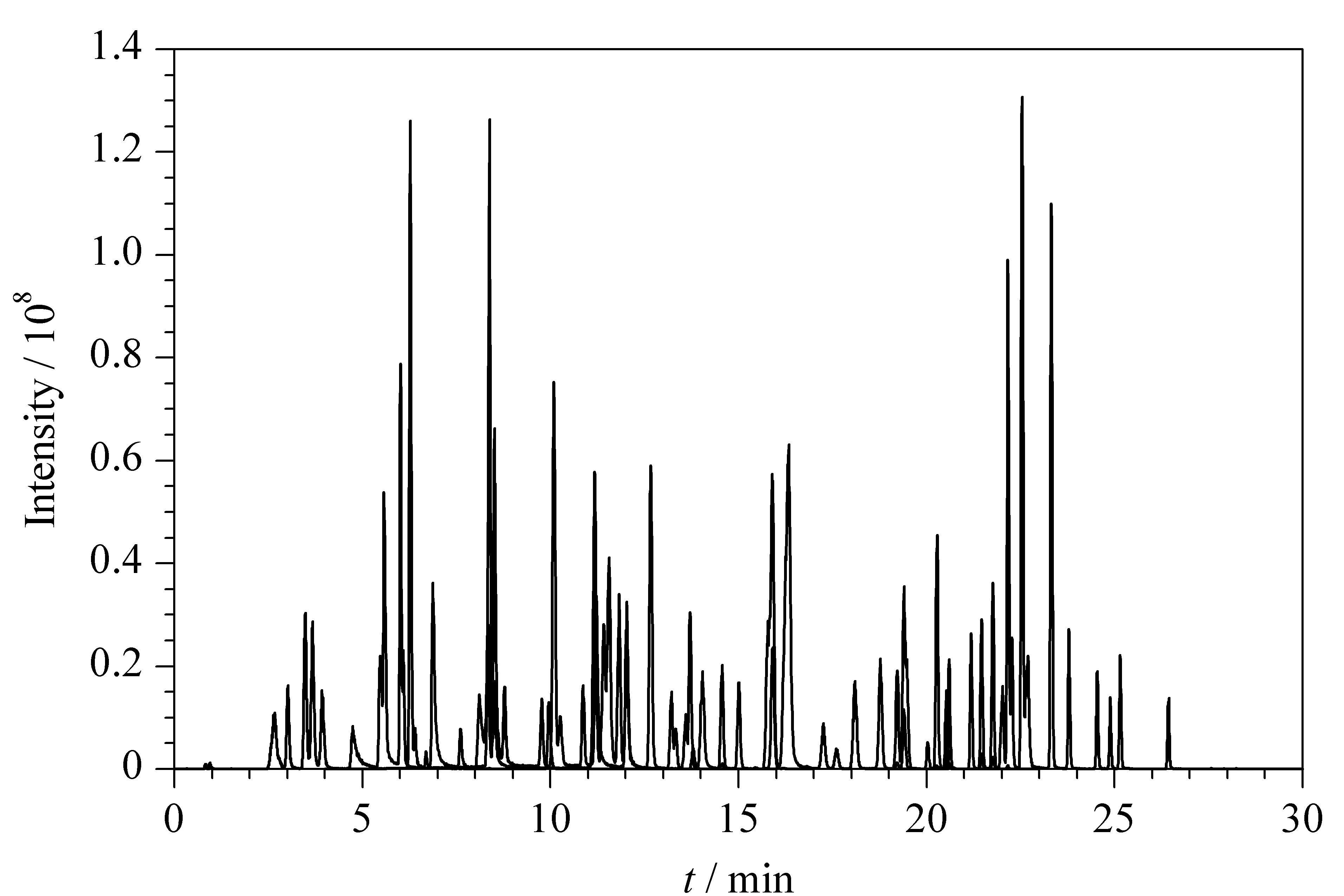
图 1 87种禁用原料混合标准溶液的总离子流色谱图Fig. 1 Total ion current chromatogram of mixed standard solution of the 87 prohibited ingredients
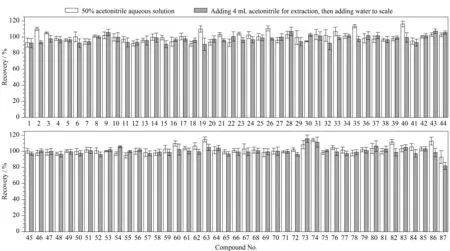
图 2 乳液膏霜类基质中87种禁用原料在不同提取溶剂下的加标回收率(n=6)Fig. 2 Spiked recoveries of the 87 prohibited ingredients with different extraction solvents in cream matrix (n=6)Compound Nos. 1-87 were the same as those in Table 1.
2.2 样品前处理条件的优化
2.2.1水基类、乳液膏霜类样品
提取溶剂的选择 本研究所涉及的87种禁用原料中,大部分禁用原料在乙腈中有较好的溶解性。经查阅相关文献[13-15,17,20-25],本研究以乳液膏霜类样品为典型基质,先加入适量1 mg/L的87种禁用原料混合标准溶液(加标水平均为2倍定量限(LOQ)),之后分别考察了用乙腈、50%乙腈水溶液、70%乙腈水溶液提取及先用乙腈提取再加水定容的4种提取方式对目标物提取效率的影响。结果显示,使用乙腈和70%乙腈水溶液提取时,由于溶剂效应,部分出峰时间较早的化合物色谱峰变宽,峰形对称性差。比较了用50%乙腈水溶液提取和先用乙腈提取再加水定容两种提取方式下目标物的加标回收率,结果如图2所示。在两种提取方式下,87种禁用原料的加标回收率分别为91.7%~116.2%和81.8%~115.7%,相对标准偏差分别为0.7%~8.4%和1.2%~8.2%。使用50%乙腈水溶液进行提取时不仅回收率更高,且操作简便、快捷,更符合化妆品快速筛查的目的,因此,选择50%乙腈水溶液作为水基类和乳液膏霜类样品的提取溶剂。
提取时间的选择 以乳液膏霜类样品为典型基质,考察了不同超声提取时间(10、20、30 min)对87种禁用原料提取效率的影响。结果表明,以50%乙腈水溶液为提取溶剂、超声提取20 min的条件下,绝大多数化合物可达最大提取效率。因此,实验选择超声提取时间为20 min。

图 3 油基类基质中87种禁用原料在不同提取溶剂下的加标回收率(n=6)Fig. 3 Spiked recoveries of the 87 prohibited ingredients with different extraction solvents in oil matrix (n=6)Compound Nos. 1-87 were the same as those in Table 1.
2.2.2油基类样品
提取溶剂的选择 根据文献[13,14]报道,本研究在空白油基类化妆品中先加入适量1 mg/L的87种禁用原料混合标准溶液(加标水平均为2倍LOQ),之后分别考察了不同体积分数(50%、70%、80%)的乙腈水溶液作为提取溶剂时对87种禁用原料提取效率的影响。结果如图3所示,使用50%乙腈水溶液提取时,丙酸睾酮和苯丙酸诺龙的加标回收率仅为49.6%和42.4%;使用70%和80%乙腈水溶液提取时,87种禁用原料的加标回收率分别为83.6%~114.9%和78.3%~115.0%,相对标准偏差分别为1.4%~9.1%和0.8%~8.0%。由此可见,70%乙腈水溶液的提取效率更高,且在该提取溶剂下出峰时间较早的化合物的峰形对称性更好。因此,选择70%乙腈水溶液作为油基类样品的提取溶剂。
提取次数的选择 实验分别考察了涡旋提取1、2、3次对87种禁用原料提取效率的影响。仅提取1次时,依美斯汀、丙酸诺龙、醋酸氯睾酮、丙酸睾酮和苯丙酸诺龙5个组分的回收率均小于80%;分别提取2次和3次时,87种禁用原料的加标回收率为83.7%~115.8%和82.0%~112.1%,相对标准偏差分别为0.9%~8.6%和0.6%~8.0%,由此可见,提取2次时已经能够满足87种禁用原料筛查测定的要求。因此,为节省检测时间,选择涡旋提取次数为2次。
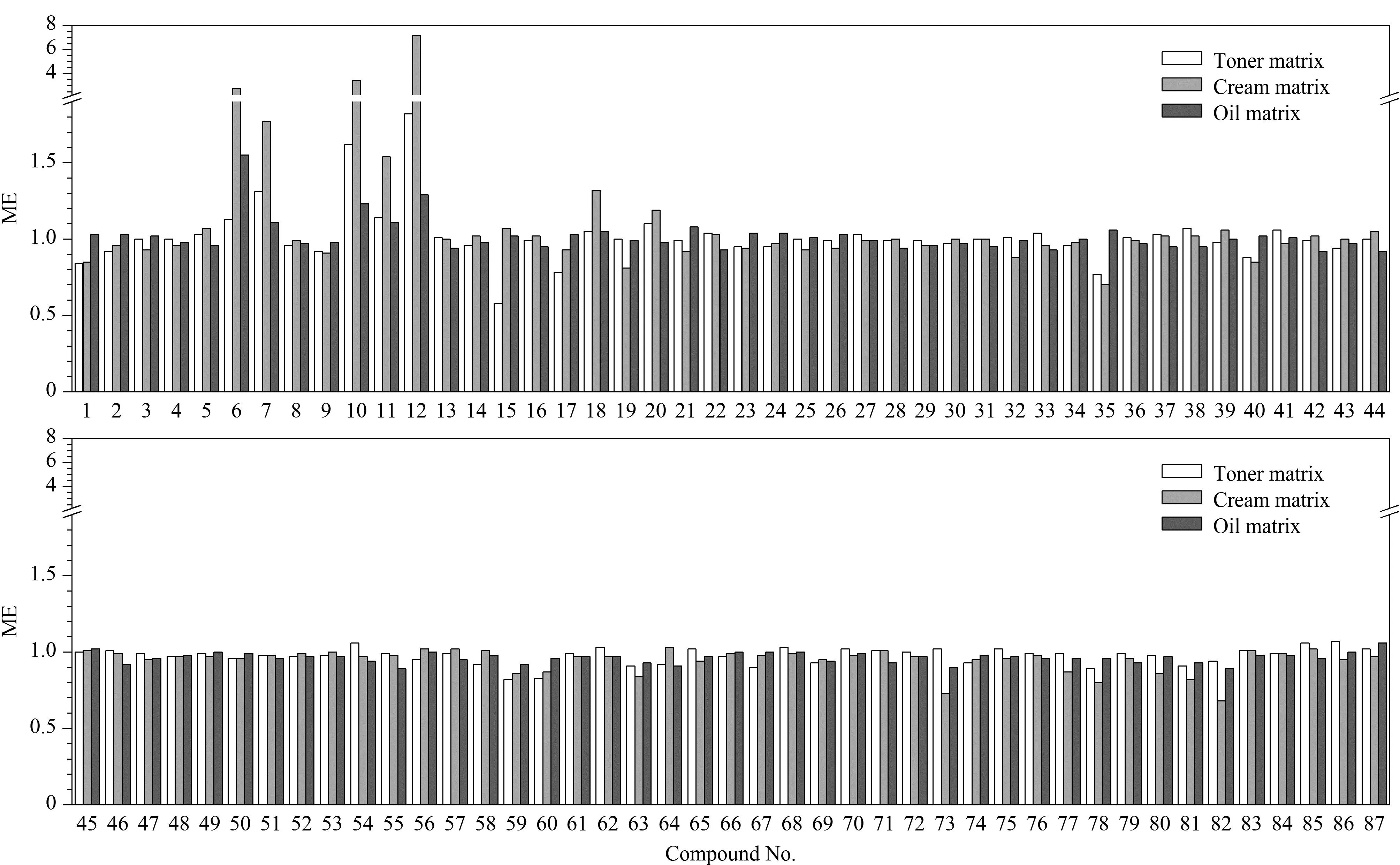
图 4 水基类、乳液膏霜类和油基类样品中87种禁用原料的基质效应Fig. 4 Matrix effects of the 87 prohibited ingredients in toner, cream and oil matricesCompound Nos. 1-87 were the same as those in Table 1.
2.3 基质效应的考察
实验分别考察了3类化妆品基质的基质效应(matrix effect, ME),并采用ME=基质匹配标准曲线斜率/溶剂标准曲线斜率的计算方法对目标化合物的基质效应进行评价;其中,ME>1表现为基质增强效应,ME<1表现为基质抑制效应;ME值越接近1,表示基质效应的影响越小,反之,则表明基质效应的影响越大[26]。实验结果表明,3类化妆品基质经溶剂提取后均有部分共萃取基质被提取,水基类、乳液膏霜类和油基类基质的ME分别为0.58~1.82、0.68~7.15和0.89~1.55(图4),均存在较明显的基质抑制或基质增强效应。因此,为了降低基质效应带来的影响,本实验采用基质匹配标准溶液进行定量分析。
2.4 线性关系、检出限及定量限
本实验利用空白基质(水基类、乳液膏霜类和油基类基质)提取液对87种禁用原料的混合标准溶液进行稀释,配制成系列质量浓度的基质匹配标准溶液。以待测物的质量浓度(x, μg/L)为横坐标、定量离子对的峰面积(y)为纵坐标,绘制基质匹配标准工作曲线。结果显示,3种化妆品基质中目标化合物在各自的线性范围内均具有良好的线性关系,相关系数(r)均大于0.99。以信噪比≥3计算检出限(LOD),以信噪比≥10计算LOQ, 87种禁用原料的LOD为0.07~0.38 μg/g,LOQ为0.21~1.15 μg/g。3种化妆品基质中87种禁用原料的线性范围、线性方程、相关系数、LOD和LOQ见表2。
2.5 回收率和精密度
在水基类、乳液膏霜类和油基类的空白样品中分别添加低、中、高(1倍LOQ、2倍LOQ、10倍LOQ)3个水平的87种禁用原料混合标准溶液,根据所建立的方法进行加标回收试验,每个加标水平重复测定6次,并计算回收率和相对标准偏差。结果如表3所示,水基类、乳液膏霜类和油基类样品中87种禁用原料的回收率分别为84.8%~115.4%、82.0%~114.9%和81.7%~112.6%,相对标准偏差分别为0.4%~9.1%、0.4%~9.1%和0.6%~9.9%。以上结果表明,所建立的化妆品中87种禁用原料的高通量检测方法准确、可靠,可用于实际样品的分析。
2.6 实际样品测定
采用所建立的方法对日常监督抽检中的349批化妆品进行分析,在8批样品中检出了禁用原料。其中,有2批样品检出甲氧苄啶,检出含量分别为53.6 mg/g和15.0 mg/g;此外,1批样品检出特比萘芬,1批样品检出萘甲唑啉,3批样品检出7-甲氧基香豆素,1批样品检出7-甲基香豆素,检出含量均大于LOD、小于LOQ。实验结果表明,非法添加《化妆品安全技术规范》(2015年版)标准方法以外的禁用原料的现象已经较为普遍。因此,本方法的建立有利于加强化妆品安全监管,为化妆品的风险监测和高通量筛查提供技术支撑。
3 结论
本研究建立了基于超高效液相色谱-串联质谱测定化妆品中8类共87种禁用原料的方法。所建立的方法操作简单、高效,定性、定量准确,适用于水基类、乳液膏霜类和油基类3类常见化妆品基质的测定。本研究在进一步规范化妆品市场、服务化妆品安全监管等方面具有较强的实用价值。
猜你喜欢
电子技术与软件工程(2021年6期)2021-11-21 01:24:53
化工设计通讯(2020年10期)2020-09-17 14:43:16
Cancer Biology & Medicine(2016年4期)2017-01-13 01:54:45
中国氯碱(2016年9期)2016-11-16 03:07:39
浙江大学学报(工学版)(2016年9期)2016-06-05 09:20:57
中学生理科应试(2016年2期)2016-05-30 14:23:28
电脑知识与技术(2016年9期)2016-05-18 11:15:21
广东石油化工学院学报(2016年3期)2016-05-17 05:16:21
服装学报(2015年1期)2015-10-21 01:20:30
现代计算机(2015年15期)2015-09-18 01:22:19
