
茯神指纹图谱结合化学模式识别和多指标成分定量研究
2024-01-09 00:46彭华胜张亚中刘军玲金传山
中草药 2024年1期
王 娜,程 璐 ,王 浩 ,彭华胜, ,张亚中 ,刘军玲 *,金传山*
1.安徽中医药大学药学院,安徽 合肥 230012
2.安徽省食品药品检验研究院,安徽 合肥 230051
3.国家药监局中药质量研究与评价重点实验室,安徽 合肥 230051
4.中国中医科学院 中药资源中心 道地药材品质保障与资源持续利用全国重点实验室,北京 100700
茯神PoriacumPiniRadix为多孔菌科真菌茯苓Poriacocos(Schw.) Wolf 菌核中间天然抱有松根的白色部分,始载于《名医别录》[1],甘、淡,平,入心、脾经,具有宁心、安神、利水功效。茯神在临床上仍是常用的大宗品种,《中国药典》1963 年版[2]曾收录“茯神”,然而,后来历版药典均未再收录[3]。全国多个省已制定中药材地方质量标准,但大多是基原、性状、功效上的描述,内容较少且不全面[4]。此外,通过查阅文献资料,发现茯神相关文献资料也较少。基于此,本研究采用高效液相色谱法[5-8]建立茯神的指纹图谱[9-10],结合相似度评价同时进行化学模式识别[11-17],并测定其中10 个三萜成分的含量。从定性和定量2 个方面为该药材质量标准的提升奠定基础,保证茯神的临床疗效与用药安全。
1 仪器与材料
Ultimate 3000 型高效液相色谱仪及所配备的二极管阵列检测器(Dionex 公司);ML 型204 万分之一电子天平(Mettler 公司);XP26 型百万分之一电子分析天平(Mettler 公司);A11 型分析研磨机(IKA公司);KQ-100 型超声仪(昆山市超声仪器有限公司);XMTD205 水浴锅(常州国宇仪器制造有限公司);Simplicity-185 型超纯水仪(Millipore 公司);HALO 90 A C18色谱柱(250 mm×4.6 mm,5 μm)
对照品16α-羟基松苓新酸(批号J12HB184946,质量分数≥99%)、茯苓酸B(批号J13HB172712,质量分数≥95% )、去氢土莫酸( 批号A19GB158345,质量分数≥95%)、茯苓酸A(批号J28HB186446,质量分数≥96%)、多孔菌酸C(批号P08J11L107514,质量分数≥95%)、3-表去氢土莫酸(批号A26IB212331,质量分数≥95%)、3-O-乙酰基-16α-羟基松苓新酸(批号P08J11S107846,质量分数≥95%)、去氢茯苓酸(批号A19IB212921,质量分数≥98%)、松苓新酸(批号P24J10S80630,质量分数≥98%)、去氢齿孔酸(批号A18IB212947,质量分数≥95%)均购于上海源叶生物科技有限公司;乙腈、甲醇为色谱纯,磷酸为分析纯,水为超纯水。
37 批茯神(S1~S17 批来自安徽,S18~S27批来自云南,S28~S37 批来自湖北),经安徽省食品药品检验研究院刘军玲主任中药师鉴定为多孔菌科真菌茯苓P.cocos(Schw.) Wolf 菌核中间抱有松根的白色部分。
2 方法与结果
2.1 色谱条件
采用HALO 90A,C18(250 mm×4.6 mm,5 μm)色谱柱,以乙腈(A)-0.05%磷酸(B)为流动相,梯度洗脱(0~9 min,62% A;9~28 min,62%~78% A;28~32 min,78%~90% A;32~38 min,90% A;38~40 min,90%~62% A),检测波长242 nm,柱温30 ℃,体积流量1.0 mL/min,进样量10 μL。
2.2 溶液的制备
2.2.1 对照品溶液的制备 精密称取16α-羟基松苓新酸、茯苓酸B、去氢土莫酸、茯苓酸A、多孔菌酸C、3-表去氢土莫酸、3-O-乙酰基-16α-羟基松苓新酸、去氢茯苓酸、松苓新酸、去氢齿孔酸对照品适量,加甲醇溶解,配制成质量浓度依次为0.038 3、0.049 0、0.048 5、0.033 7、0.047 0、0.043 0、0.038 4、0.054 7、0.089 6、0.040 9 mg/mL 的混合对照品溶液。
2.2.2 供试品溶液的制备 取本品粉末约1 g,精密称定,置具塞锥形瓶中,精密加入甲醇25 mL,超声处理(功率250 W、频率40 kHz)30 min,滤过,滤液蒸干,残渣用甲醇定容至5 mL 量瓶中,摇匀,滤过,即得。
2.2.3 阴性对照品溶液的制备 取甲醇,作为阴性对照品溶液。
2.3 HPLC 指纹图谱方法学考察
2.3.1 专属性试验 取混合对照品溶液、供试品溶液(S2)和阴性对照溶液(甲醇溶液)分别按“2.1”项下色谱条件进样测定。结果显示,在色谱图中各待测成分峰分离度良好,溶剂对色谱峰无影响,见图1。

图1 茯神供试品 (A)、混合对照品 (B) 和阴性对照品 (C)溶液的色谱图Fig.1 Chromatograms of Poria cum Pini Radix test substance (A), mixed control substance (B), and negative control substance (C)
2.3.2 精密度试验 取茯神样品(S2),按照“2.2.2”项方法制备供试品溶液,按“2.1”项色谱条件重复进样6 次,计算得到各共有峰相对保留时间及相对峰面积的RSD 均<1.5%,表明仪器精密度良好。
2.3.3 重复性试验 取茯神样品(S2),按照“2.2.2”项方法平行制备6 份供试品溶液,按照“2.1”项色谱条件进样分析,计算得到各共有峰相对保留时间及相对峰面积的RSD 均<1.0%,表明该方法重复性良好。
2.3.4 稳定性试验 取茯神样品(S2),按照“2.2.2”项方法制备供试品溶液,按“2.1”项色谱条件于制备后的0、2、4、6、12、24 h 进样,计算各共有峰相对保留时间及相对峰面积RSD 均<2.0%,表明供试品溶液在24 h 内稳定。
2.4 指纹图谱的建立及分析
2.4.1 指纹图谱的建立 将所得的37 批茯神指纹图谱依次导入到“中药色谱指纹图谱相似度评价软件(2012 版)”。以样品S1 作为参照图谱,时间窗宽度设为0.1,采用中位数法,利用多点校正法进行峰匹配,生成对照图谱,37 批茯神色谱图与对照图谱的叠加图谱见图2。

图2 37 批茯神HPLC 叠加图谱 (S1~S37)及对照图谱(R)Fig.2 HPLC overlay spectra (S1−S37) and control spectra(R) of 37 batches of Poria cum Pini Radix
2.4.2 指纹图谱的分析 37 批茯神指纹图谱标定10 个共有峰,通过与对照品比对,指认出1 号峰为16α-羟基松苓新酸、2 号峰为茯苓酸B、3 号峰为去氢土莫酸、4 号峰为茯苓酸A、5 号峰为多孔菌酸C、6号峰为3-表去氢土莫酸、7号峰为3-O-乙酰基-16α-羟基松苓新酸、8 号峰为去氢茯苓酸、9 号峰为松苓新酸、10 号峰为去氢齿孔酸。计算37 批不同产地茯神之间的相似度,见表1,结果显示,除S23、S37 外,其余35 批茯神样品的相似度在0.791~1.000,S23、S37 可能由个体差异导致,其余样品相似度较高。

表1 37 批茯神相似度评价结果Table 1 Similarity evaluation results of 37 batches of Poria cum Pini Radix
2.5 多指标成分定量
2.5.1 线性关系考察 精密称定16α-羟基松苓新酸、茯苓酸B、去氢土莫酸、茯苓酸A、多孔菌酸C、3-表去氢土莫酸、3-O-乙酰基-16α-羟基松苓新酸、去氢茯苓酸、松苓新酸、去氢齿孔酸的对照品适量,以甲醇配制成6 个不同质量浓度的系列混合对照品溶液,按“2.1”项下色谱条件进样测定峰面积。以对照品浓度为横坐标(X),对照品峰面积为纵坐标(Y)进行线性回归分析,结果显示各组分在相应浓度范围内线性关系良好。线性回归方程、线性范围及相关系数见表2。

表2 茯神中10 种成分的线性回归结果Table 2 Linear regression results of 10 components in Poria cum Pini Radix
2.5.2 精密度试验 精密吸取“2.2.1”项下的混合对照品溶液,按“2.1”项下色谱条件连续进样6 次,记录峰面积,结果16α-羟基松苓新酸、茯苓酸B、去氢土莫酸、茯苓酸A、多孔菌酸C、3-表去氢土莫酸、3-O-乙酰基-16α-羟基松苓新酸、去氢茯苓酸、松苓新酸、去氢齿孔酸峰面积的RSD分别为0.001 6%、0.001 1%、0.001 9%、0.002 6%、0.003 9%、0.002 0%、0.001 7%、0.001 6%、0.002 4%、0.002 4%,表明仪器精密度良好。
2.5.3 重复性试验 精密称取茯神样品粉末6 份(S2),按照“2.2.2”项方法制备供试品溶液,在“2.1”项色谱条件下平行进样分析,结果16α-羟基松苓新酸、茯苓酸B、去氢土莫酸、茯苓酸A、多孔菌酸C、3-表去氢土莫酸、3-O-乙酰基-16α-羟基松苓新酸、去氢茯苓酸、松苓新酸、去氢齿孔酸质量分数的RSD 分别为0.002 8%、0.001 1%、0.001 4%、0.001 7%、0.013 4%、0.001 4%、0.001 5%、0.001 3%、0.001 1%、0.002 7%,符合实验要求。
2.5.4 稳定性试验 精密吸取供试品溶液(S2),在“2.1”项色谱条件下,分别于0、2、4、6、12、24 h 进样,计算出含量,结果16α-羟基松苓新酸、茯苓酸B、去氢土莫酸、茯苓酸A、多孔菌酸C、3-表去氢土莫酸、3-O-乙酰基-16α-羟基松苓新酸、去氢茯苓酸、松苓新酸、去氢齿孔酸质量分数的RSD 分别为0.002 3%、0.000 8%、0.000 6%、0.001 1%、0.017 2%、0.000 9%、0.001 5%、0.003 0%、0.000 7%、0.001 6%,样品稳定性良好,符合实验要求。
2.5.5 加样回收试验 精密称取已知成分含量的茯神样品(S2)0.5 g 置锥形瓶中,平行6 份,按1∶1 比例加入各对照品,按“2.2”项下方法制备供试品溶液,在“2.1”项色谱条件下进样分析,计算得到16α-羟基松苓新酸、茯苓酸B、去氢土莫酸、茯苓酸A、多孔菌酸C、3-表去氢土莫酸、3-O-乙酰基-16α-羟基松苓新酸、去氢茯苓酸、松苓新酸、去氢齿孔酸的平均加样回收率分别为 90.27%、109.22%、97.90%、85.57%、107.41%、86.25%、88.45%、108.60%、96.22%、85.22%,RSD 分别为0.033 4%、0.016 1%、0.026 4%、0.024 6%、0.032 7%、0.029 7%、0.026 1%、0.030 3%、0.049 8%、0.036 8%,表明本方法的加样回收率良好。
2.5.6 样品含量测定 取37 批茯神药材粉末各约1.0 g,精密称定,按“2.2”项下方法制备供试品溶液,按“2.1”项下色谱条件测定,计算16α-羟基松苓新酸、茯苓酸B、去氢土莫酸、茯苓酸A、多孔菌酸C、3-表去氢土莫酸、3-O-乙酰基-16α-羟基松苓新酸、去氢茯苓酸、松苓新酸、去氢齿孔酸10个成分的含量。结果见表3。
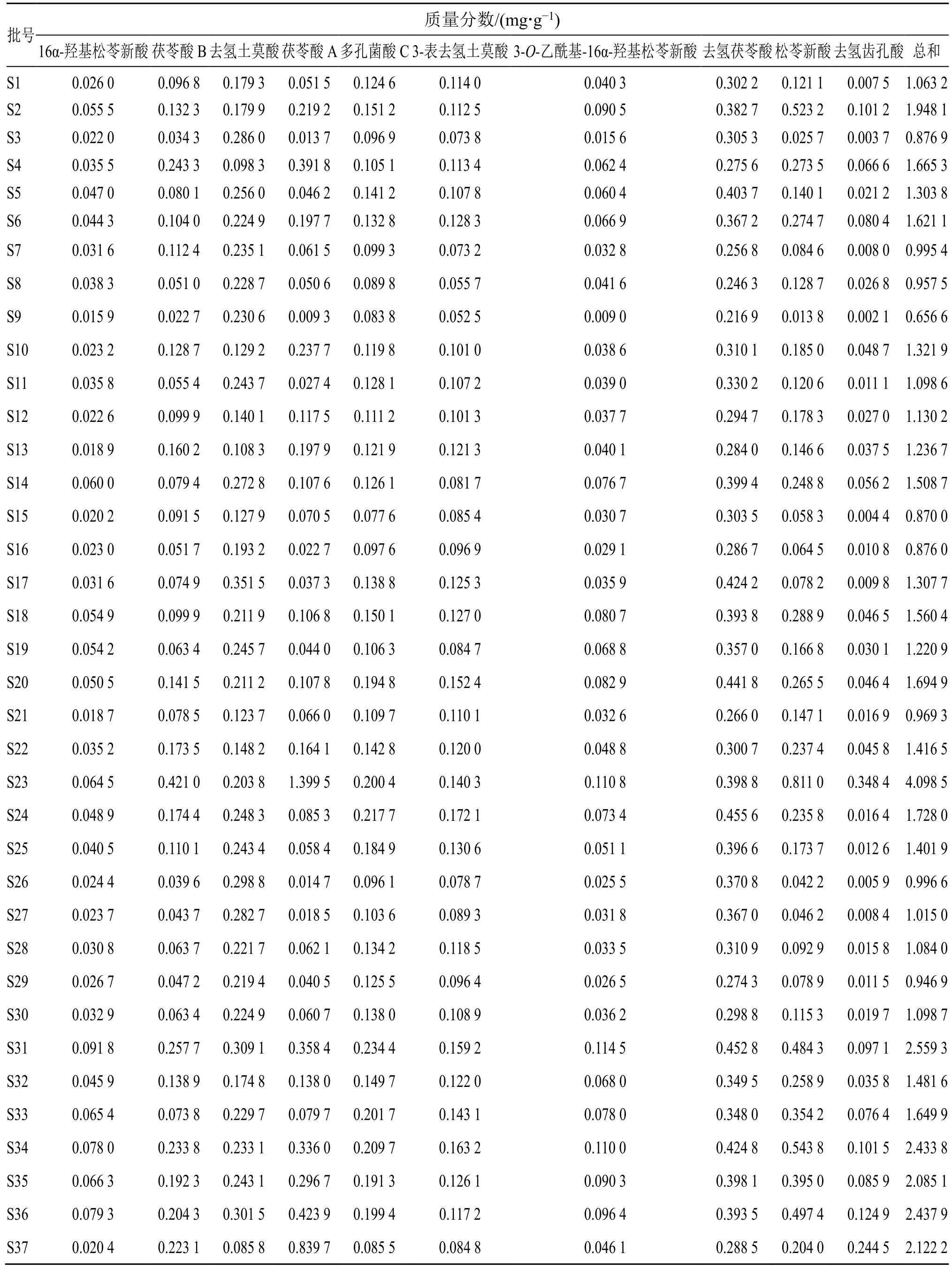
表3 样品含量测定结果Table 3 Results of content determination of samples
2.6 化学模式识别分析
2.6.1 聚类分析 以37批茯神中10种化学成分的含量为变量,以平方欧氏距离为测距,运用IBM SPSS Statistics 23 软件,采用系统聚类法,对37批茯神进行聚类分析,结果见图3。当平方欧氏距离为5 时可分为3 类,S23 聚为一类,S37 聚为一类,其余样品聚为一类,S23、S37 可能由个体含量差异导致,其余样品间质量差异不大,与相似度结果相符。

图3 37 批茯神样品的聚类分析图Fig.3 Cluster analysis of 37 batches of Poria cum Pini Radix samples
2.6.2 主成分分析(principal component analysis,PCA) 为进一步比较不同批次茯神间的质量差异,将37 批茯神10 个有效成分含量导入IBM SPSS Statistics 23 和Simca 软件,进行PCA,共有峰特征值见表4。前2 个主成分的初始特征值依次为6.098、2.248,累积方差贡献率达到83.460%,表明以上2个主成分能够较好的代表指纹图谱中的大部分信息。PCA 得分图显示37 批样品可大致划分为3 类,与CA 结果一致,见图4。

表4 主成分特征值及方差贡献率Table 4 Principal component eigenvalues and variance contribution rate
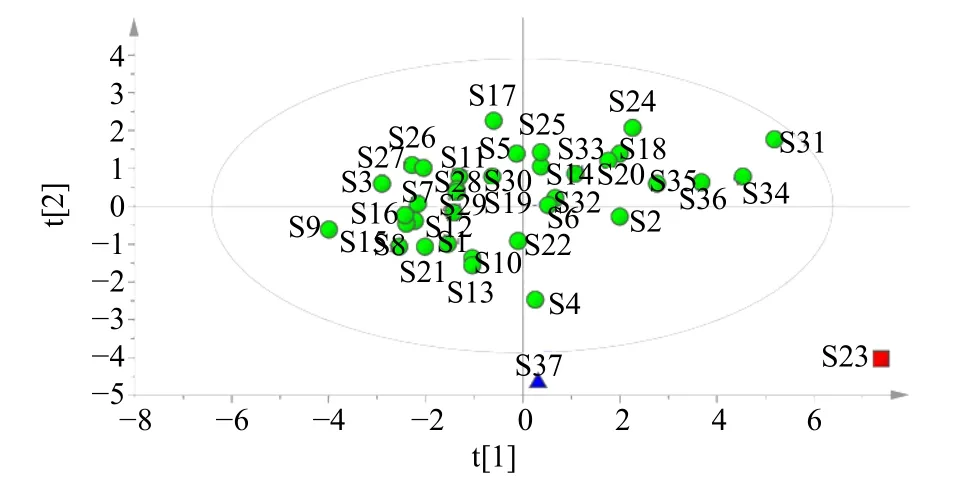
图4 37 批茯神PCA 得分图Fig.4 PCA score chart of 37 batches of Poria cum Pini Radix
2.6.3 正交偏最小二乘判别分析(orthogonal partial least squares discriminant analysis,OPLS-DA) 将37 批茯神有效成分含量数据导入到Simca 软件进行OPLS-DA,以变量重要性投影(variable importance in projection,VIP)值>1.0 为标准进行筛选,结果见图5,表明共有4 个特征峰的VIP 值>1.0,分别为峰8 去氢茯苓酸(VIP=1.775 9)、峰1 为16α-羟基松苓新酸(VIP=1.333 9)、峰5 为多孔菌酸C(VIP=1.125 1)、峰6 为3-表去氢土莫酸(VIP=1.107),表明这4 个成分是茯神不同批次样品间质量差异贡献较大的标志物,可以用来区分不同产地、批次间茯神的质量差异。OPLS-DA 分析的结果与CA、PCA 的结果一致。
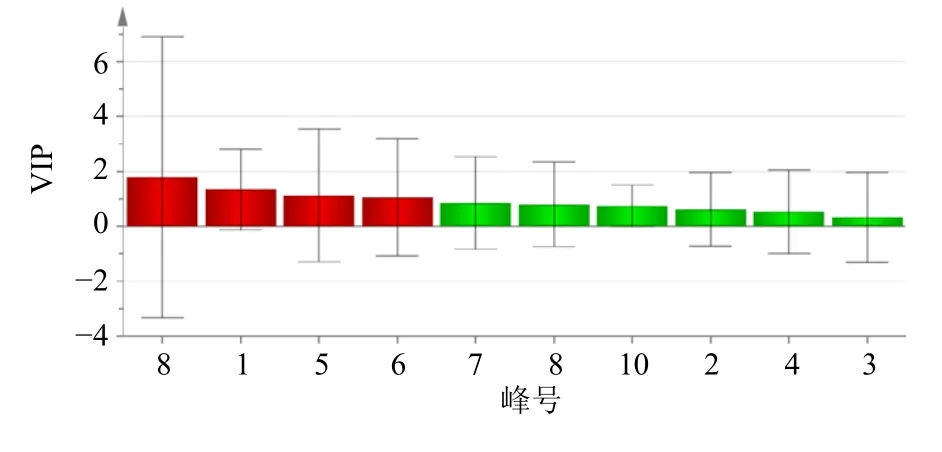
图5 37 批茯神OPLS-DA 模型VIP 值图Fig.5 VIP value chart of 37 Batch of Poria cum Pini Radix
3 讨论
3.1 色谱条件及处理方法的选择
本实验在实验操作过程中选用高效液相色谱法测定茯神三萜类成分,所用提取溶剂有乙醇、甲醇,提取方法有超声与加热回流,提取时间有20、30、40 min,发现甲醇超声30 min 后提取的样品溶液色谱峰信息较准确完整、操作简单且高效节约,适用于茯神的定性及定量研究。在波长的选择上,本文根据10 种待测成分的吸收波长特点,分别考察了在210、242 nm 波长下的检测效果。结果发现,在210 nm 波长下10 种三萜类成分均无吸收,在242 nm 波长下有最大吸收,故最终选择242 nm 为测定波长。另外对流动相亦进行了考察,有乙腈-0.05%磷酸、乙腈-0.1%磷酸等。结果表明,以乙腈-0.05%磷酸进行梯度洗脱获得的图谱峰形良好,基线分布稳定,可分别满足对10 种三萜类化合物图谱的分离要求。
3.2 指纹图谱的建立
本实验通过对37 批茯神药材在相同的提取方法和色谱条件下所得HPLC 图谱及数据进行分析,结合中药色谱指纹图谱相似度评价软件(2012 版)分析结果,确定了茯神中共有15 个共有色谱峰,比对对照品色谱峰指认了16α-羟基松苓新酸、茯苓酸B、去氢土莫酸、茯苓酸A、多孔菌酸C、3-表去氢土莫酸、3-O-乙酰基-16α-羟基松苓新酸、去氢茯苓酸、松苓新酸、去氢齿孔酸,且在37 批样品中均检出这10 个成分。此外,部分样品在26 min 左右检出1个未知峰,这可能与茯神木的成分有关,有待进一步研究。除S23、S37 外,其余35 批茯神样品的相似度在0.791~1.000,S23、S37 两批样品相似度存在较大差异,可能由个体差异导致,其余样品相似度较高,表明茯神多批次产地的质量相对稳定,能很好地反应样品的指纹图谱。
3.3 含量测定及化学计量学分析
本实验通过CA、PCA 及OPLS-DA 分析对茯神含量进行综合分析。通过CA 发现,37 批茯神样品可聚为3 类,S23 聚为一类,S37 聚为一类,其余样品聚为一类,与相似度结果相符。对比表3 含量结果图发现,S23 中大多成分含量较高,而S37中大多成分的含量均较低,表明茯神中某些成分含量的高低可能是这两批样品各单独聚为一类的原因,产地来源并不是决定聚类结果的唯一因素;PCA得分图与CA 结果一致,利用主成分分析得到2 个主成分,可代表样品中的大部分信息;此外,OPLS-DA 筛选出去氢茯苓酸、16α-羟基松苓新酸、多孔菌酸C、3-表去氢土莫酸这4 个质量差异标志物,表示这4 个色谱峰所代表的成分对区分不同产地、批次茯神的贡献较大,可以用来区分不同批次间茯神的质量差异。
4 结语与展望
茯神是卫生部颁布的第一批药食同源两用品[18],仅收载于地方标准,而且标准过于简单,无法有效控制茯神质量,本研究建立的指纹图谱及含量测定方法重复性良好,准确度高,可为茯神的质量控制提供新方法,弥补了现有质量标准的不足,可用于茯神的质量评价。此外,筛选出影响茯神质量的4 个潜在差异标志物,可为进一步完善该药材的质量控制标准提供参考。
中药材作为中医用药的源头,其质量控制对后续中药饮片和中成药的安全性、有效性起到关键性作用,进而影响其临床疗效。通过市场调研,了解到目前市场流通的茯神有部分为后期在茯苓中人为插入松根而成,只为满足茯神外观性状要求,而非真正的抱木而生,不具备茯神的功效;另外,以松根为中心,靠近松根多远的范围,算作真正意义上的茯神,暂无统一的标准,有待进一步研究,以确定其规格等级。因此,需进一步完善茯神质量标准体系,建立科学、管用的质量标准,服务于监管,同时解决茯神质量掺伪问题。
利益冲突所有作者均声明不存在利益冲突
猜你喜欢
江西中医药(2022年10期)2022-10-25
基层中医药(2020年7期)2020-09-11
妇女生活(2019年8期)2019-08-12
基层中医药(2018年10期)2018-12-06
中成药(2018年2期)2018-05-09
中成药(2018年3期)2018-05-07
新乡学院学报(2016年6期)2016-12-01
保健与生活(2016年1期)2016-04-12
当代化工研究(2016年9期)2016-03-20
